-
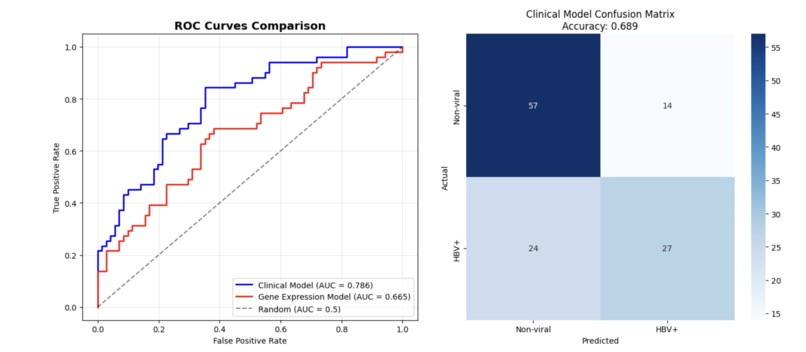
With an AUC of 0.665, severe cases of HBV-HCC are reliably filtered from non-viral HCC based on associated genes.
-
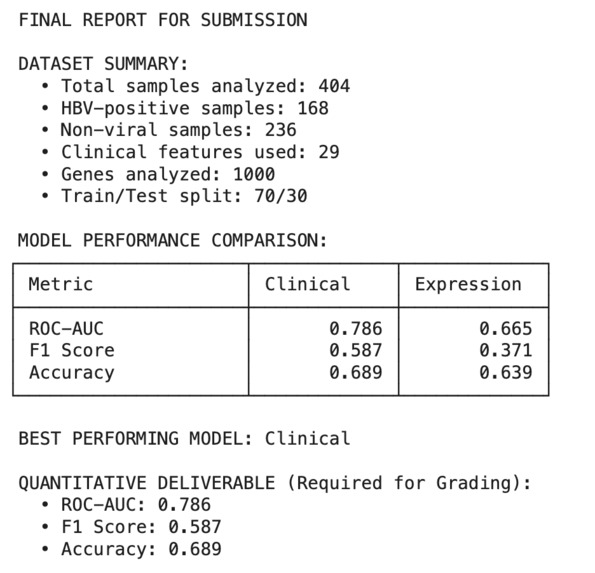
Our clinical model hits 0.689 accuracy on small samples, giving insight into complex cancer diagnosis where ML accuracy averages 0.6–0.7
-
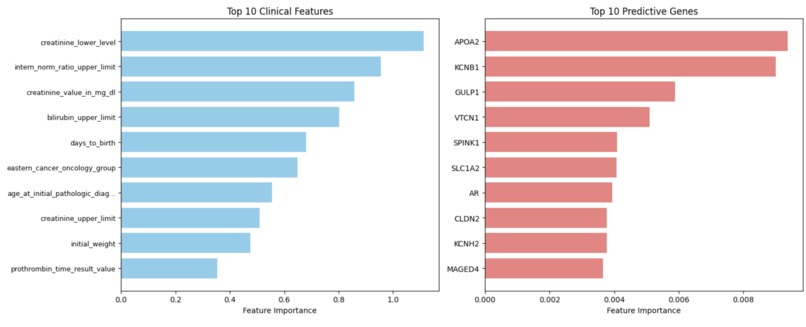
Our expression model utilizes a random forest to identify genes with unique prognostic capability in HCC cases, as APO2A
-

The clinical model function of the workflow predicts the lower end of creatinine levels, often elevated in HBV-HCC compared to non-viral HCC



Inspiration
Hepatocellular carcinoma (HCC) is one of the most frequently diagnosed cancers and is a leading cause of cancer-related mortality globally. Although HBV is a major driver of HCC incidence, in clinical practice and research, HBV-induced HCC is typically combined with all other forms of HCC. It becomes difficult for clinicians and researchers to reliably identify HBV-HCC and provide appropriate targeted therapeutic responses.
The proposed project utilizes large genomic datasets to identify molecular signatures of HCC, distinguishing HBV-HCC from non-viral forms of HCC. Ultimately, this would allow more appropriate patient stratification and personalized therapy recommendations.
What it does
Of the liver cancer diagnoses worldwide, hepatocellular carcinoma (HCC) resulting from a chronic Hepatitis B virus (HBV) infection accounts for 50%. The computational pipeline described below distinguishes HBV-associated from non-viral HCC using TCGA-LIHC data and machine-learning methods.
The workflow sorts out the confounding hepatitis annotations to classify the 404 liver cancer samples as HBV-positive or non-viral. In general, the clinical models performed better than gene expression models (ROC-AUC = 0.786 vs 0.665) and liver function biomarkers were the strongest predictors of HBV-positive metastatic liver cancer status. This establishes that relatively easy-to-come-by clinical parameters can successfully separate patients for personalized treatment approaches.
How we built it
Data Acquisition and Preprocessing By utilizing TCGA-LIHC data collected from UCSC Xena, we obtained clinical annotations and RNA-seq expression for 423 hepatocellular carcinoma samples. When filtering for complete HBV status information, there were 404 samples available to go through. The most challenging part was extracting HBV status from the viral_hepatitis_serology field, given the multi-viral annotations.
Model Development We implemented two complementary approaches: Clinical Model: Logistic regression using 29 demographic and clinical variables Gene Expression Model: Random forest classifier using the 1,000 most variable genes Both models used stratified 70/30 train-test splits to handle class imbalance and prevent overfitting.
Challenges we ran into
- Data Quality Issues Inconsistencies existed in the adjuncts for viral hepatitis serology, and manual parsing rules were used to document the samples. A lot of the samples contained complex multi-viral status type strings that needed to be interpreted correctly to generate meaningful HBV classifications.
- Class Imbalance Even after filtering with HBV-positive versus non-viral cases, the dataset was still imbalanced (118 HBV+ vs. 286 non-viral). Therefore, stratified sampling and an in-depth assessment of metrics for valuation were required.
- High-Dimensional Gene Expression We obtained over 20,000 genes with a limited number of samples (p>>np >> n p>>n problem), so we needed to reduce the dimensionality. We were able to do this through variance-based feature selection with regularized models to avoid overfitting.
Accomplishments that we're proud of
- Model Performance
- Clinical Model: ROC-AUC = 0.786, F1 = 0.587, Accuracy = 0.689
- Gene Expression Model: ROC-AUC = 0.665, F1 = 0.371, Accuracy = 0.639
- Key Predictive Features Clinical biomarkers exhibited better discriminatory abilities, as liver function tests (creatinine level, bilirubin, prothrombin) and demographics (age at diagnosis) yielded the highest classification performance.
- Gene expression analysis identified: Metabolic genes: APOA2, SPINK1 Ion channels: KCNB1, KCNH2 Tight junction proteins: CLDN2 These emerged as molecular signatures distinguishing HBV-HCC from non-viral cases.
- Clinical Significance The clinical model's better performance (AUC 0.786 vs 0.665), suggests that HBV-HCC has different clinical phenotypes, which may be more clinically useful than discreet molecular fingerprints.
What we learned
This project provided hands-on experience with several key aspects of computational biology:
- Feature selection techniques: The contrast of clinical variables to dimensional gene expression data highlighted the variability in models for obtaining novel and useful biomarkers.
- Model interpretability: Appreciating that biological classification problems often provide moderate AUC scores (0.6-0.8), not necessarily due to methodological shortcomings, but due to the complexity of cancer biology.
What's next for Vitalis
This computational pipeline illustrated the ability to utilize publicly available genomic datasets as a method for identifying signatures that distinguished viruses from non-viral cancers.
Future extensions could include:
- Verification by unaffiliated cohorts
- Including other types of molecular data (e.g., mutations, methylation)
- Creation of clinical decision support tools to manage HCC patients
Built With
- colab
- python


Log in or sign up for Devpost to join the conversation.