Inspiration
🔬 Why Do We Care About Protein Expression?
The phenotype of a cell is demarcated by its proteins. Although RNA measurements are correlated with protein, we often don't fully understand the fundamental biology between the steps of a mature RNA molecule and a protein being expressed and used in the cell. Proteins are difficult to capture and quantify at a high-throughput level (1). Several biological phenomena are critical in this respect - protein expression marks changes in our cells during development and in many disease processes such as cancer. Understanding the links with RNA and reliably measuring more surface and cytoplasmic proteins remains a major challenge (1).
🧪 CITE-seq: A Step Towards Unified Benchmarking
Predicting protein expression is an open challenge with limited data because high throughput protein sequencing is not readily available. Large CITE-seq datasets are a step in this direction, combining measurements of both RNA expression and surface protein abundance at the single cell level.
🧬 The Protein Prediction Puzzle
Surface protein abundance is influenced by a wide breadth of factors such as transcription, translation, protein localization, cell cycle, and cell-cell signalling. Previous studies have determined that RNA expression alone is not sufficient to reliably predict protein expression.
💡 Enter the Biological Foundation Models
We hypothesize that biological foundation models can help bridge this gap by capturing protein properties encoded in RNA- and protein- sequence (2,3). Their pre-trained embeddings can help the model generalize in this challenging data constrained setting.
Accomplishments that we're proud of
📊 Evaluating Mutual Information
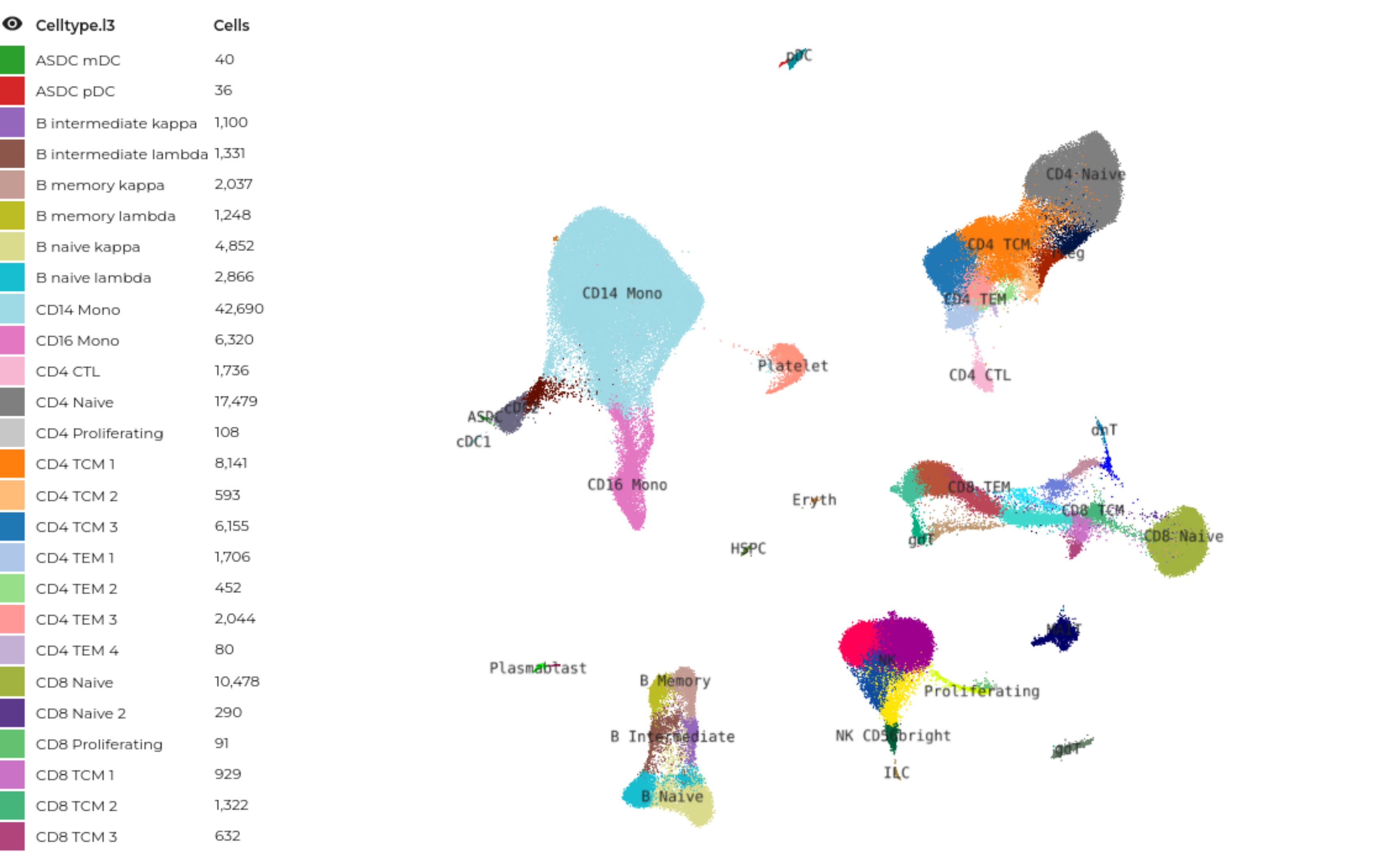
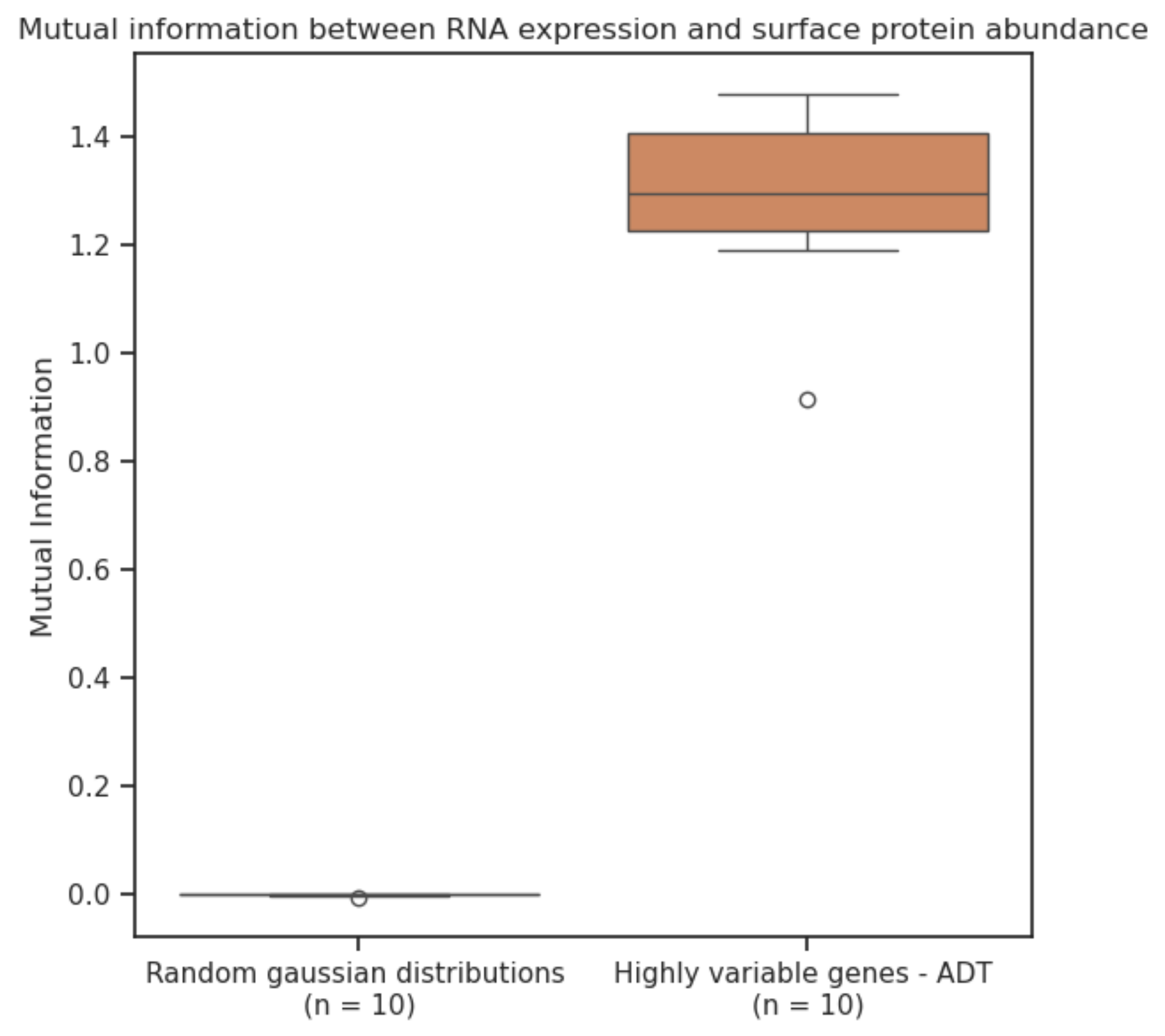
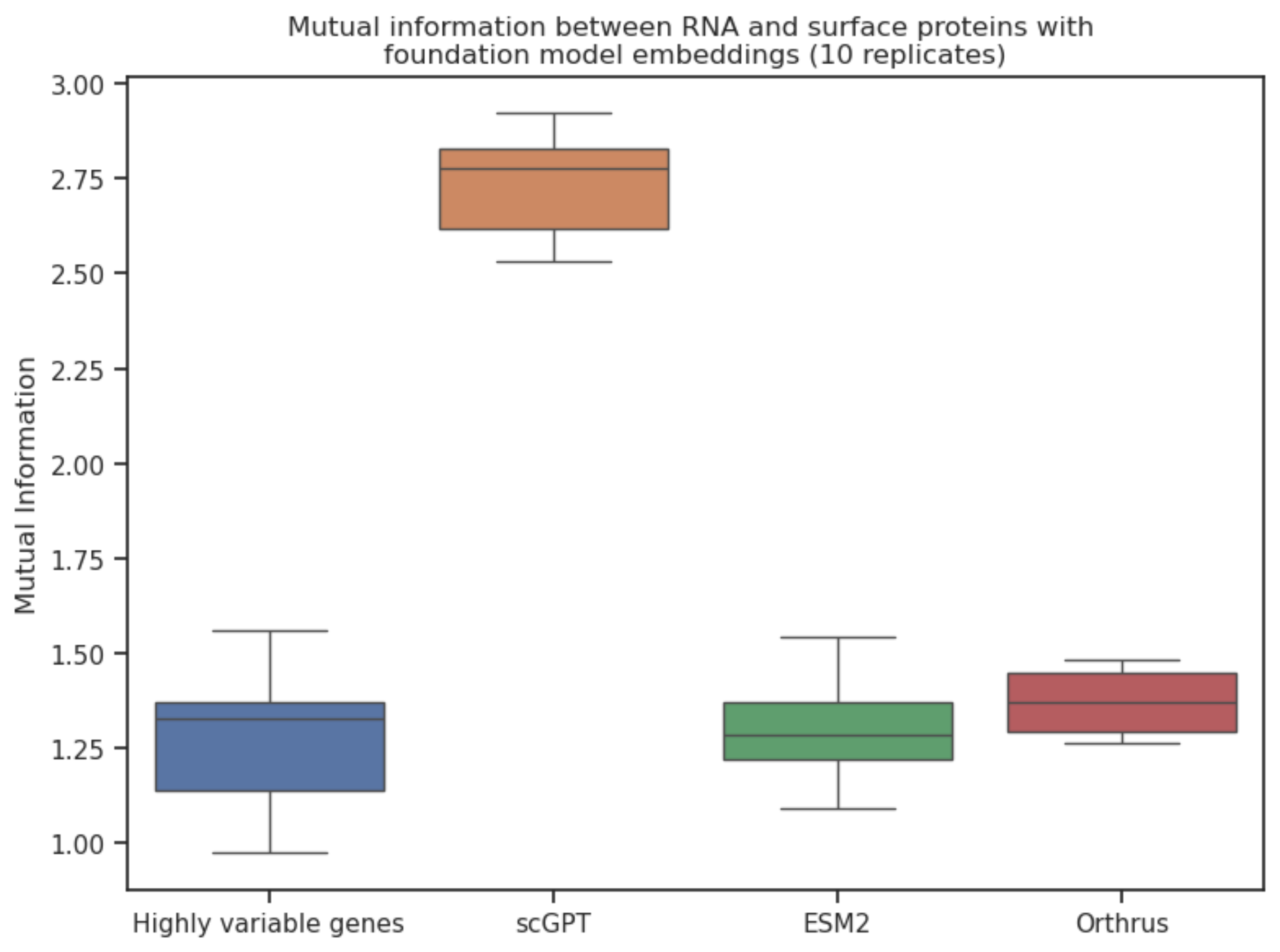
We evaluate the mutual information (4) between RNA expression and surface protein abundance through a large CITE-seq dataset of peripheral blood mononuclear cells (PBMCs) and find that scGPT embeddings (5) are more informative than a baseline of 2000 highly variable genes. This is a novel way of thinking about the amount of information one modality captures about another one.
🔍 A Unique Cross-Modality Perspective
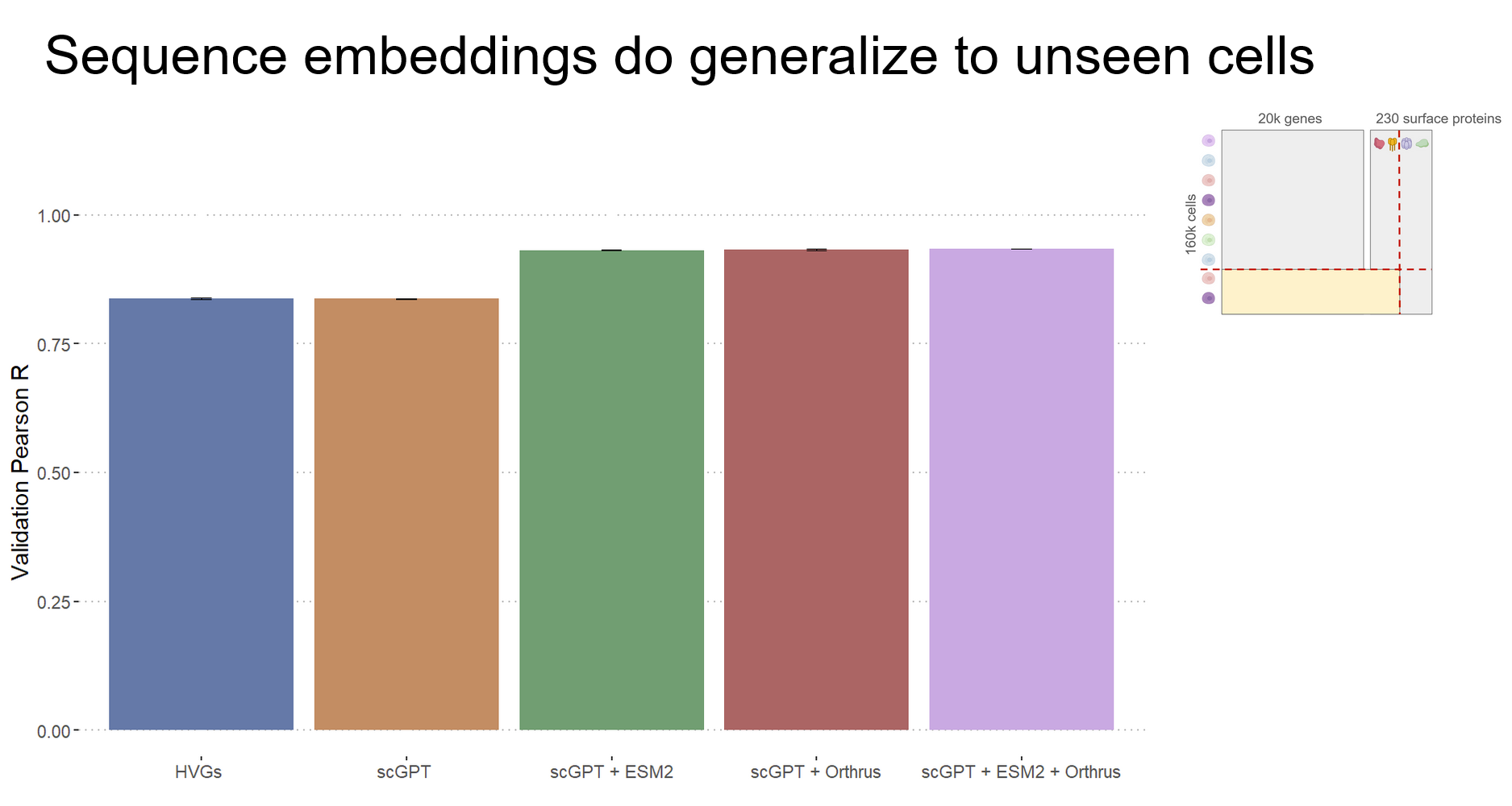
We evaluate the utility of cell model scGPT (5) sequence models ESM2 (3) and Orthrus (4) foundation models on CITE-seq data to determine if zero-shot representations in these models can more accurately predict protein abundance from RNA expression. Our analysis offers a unique perspective on this prediction task by determining real bounds on mutual information and evaluating the performance of cutting-edge foundation models on this unique task, which allows us to for the first time, directly compare the performance of foundation models designed across different modalities: RNA expression, RNA sequence, and protein sequence.
💪 A Benchmark for Surface Protein Expression Prediction
Through our experiments, we discovered that predicting protein expression, especially on unseen proteins is a very difficult task. We benchmarked both sequence-based and cell-context-based foundation models for this task, and our framework will allow for method-developers to test their pretrained foundation models to see if the additional context will allow for increases in performance on this task. We plan to package our scripts into a complete end-to-end pipeline for benchmarking of the outlined tasks.
Challenges we ran into
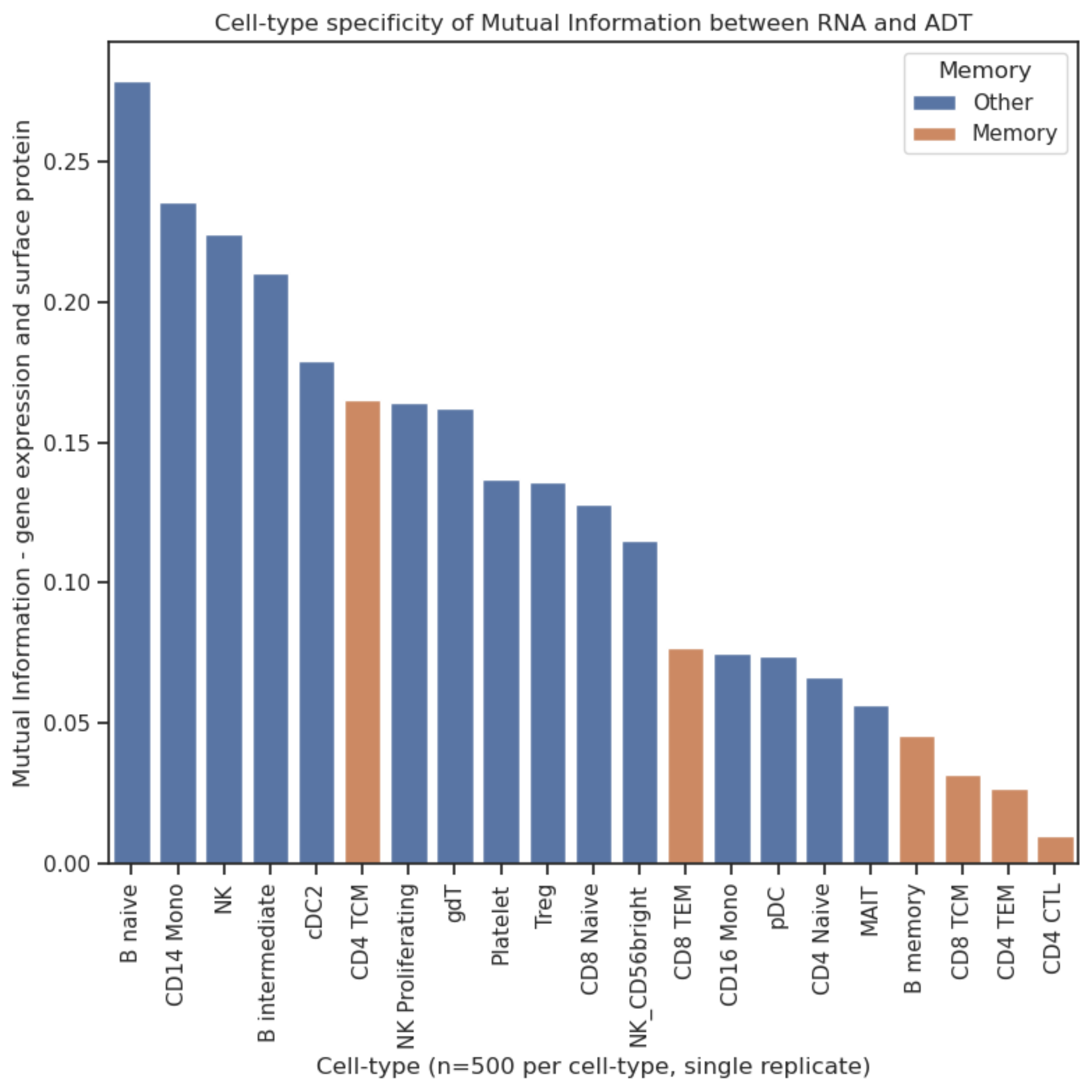
We expected that providing sequence level embeddings will allow our models to generalize to new cite seq experimental data. We find that the answer is a bit more nuanced than that. We are indeed able to improve generalization to new cells for proteins that the models have previously seen. However, for the more challenging task of generalizing to new proteins, we find that providing sequence embeddings does not improve performance. This was surprising to us since our intuition was that providing sequence level information would allow us to predict new previously unseen sequences. Our conclusion was that from 152 protein sequences there is simply not enough diversity for us to learn a reliable function to predict protein expression, and so we would need more data to succeed at this task. We found it hard to avoid over-fitting, even with strong regularization!
What we learned
There's still more signal left to capture in order to predict surface protein abundance. Higher mutual information didn't necessarily correspond to improved predictive performance. This might be because of model misspecification or incomplete optimization, but it does lead us to conclude that we are missing something and this will be an important future research direction.
References
Peterson, V. M. et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 35, 936–939 (2017).
Lin, Z. et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction.
Fradkin, P., Shi, R., Wang, B., Frey, B. & Lee, L. J. Splicing Up Your Predictions with RNA Contrastive Learning. Preprint at http://arxiv.org/abs/2310.08738 (2023).
Gowri, G., Lun, X.-K., Klein, A. M. & Yin, P. Approximating mutual information of high-dimensional variables using learned representations. Preprint at http://arxiv.org/abs/2409.02732 (2024).
Cui, H. et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat Methods 21, 1470–1480 (2024).

Log in or sign up for Devpost to join the conversation.