-
-
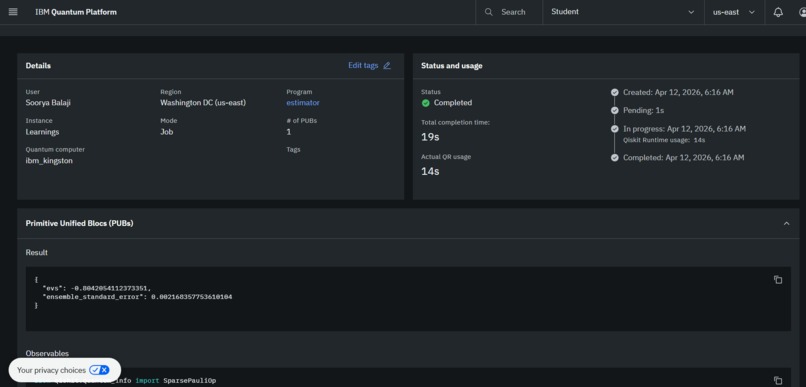
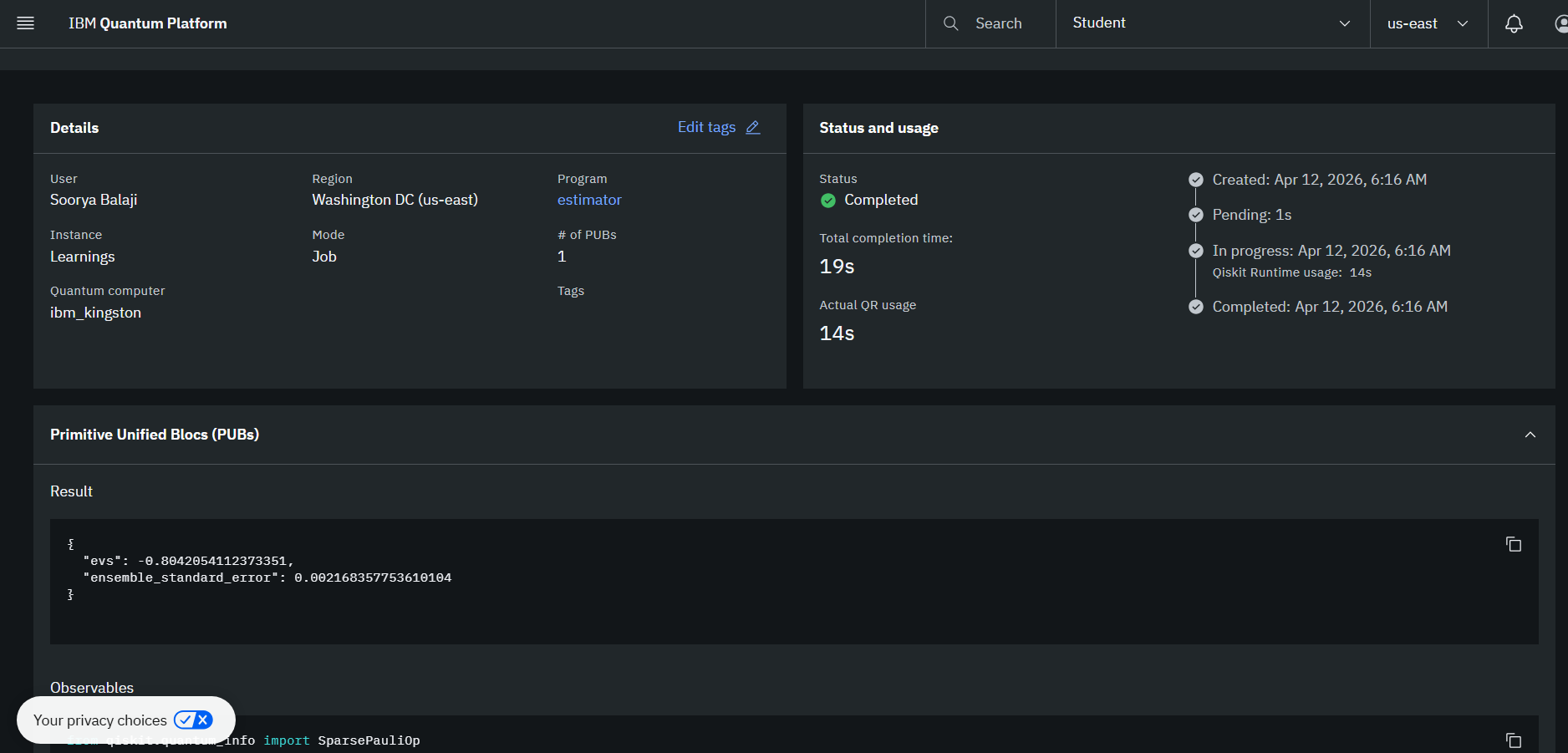
Real execution on an IBM Quantum Computer
Inspiration
We are currently living through the revolution of Quantum Computing. For years, quantum chemistry has been nothing but a theoretical field, something experimental, and restricted to models in academic papers. I built QuantumBind because I wanted to take these abstract algorithms, such as VQE (Variational Quantum Eigensolver), out of the lab and and apply them to one of the biggest and most expensive bottlenecks of modern science and medicine: Drug Discovery. I built QuantumBind because I wanted to advance the capabilities of quantum chemistry, see how we could take these revolutionary ideas and figure out how to use them to solve one of the most expensive and pressing issues of our time.
Modern drug development is a game of trial and error. It currently takes an average of 10 years and $2.6 billion to bring a single drug to market, mainly because classical computers don't have the power to simulate subatomic electron correlations with good enough accuracy. A drug might look like a good fit classically, but at the quantum level, the electron clouds might repel each other just enough to render the drug useless.
But we're sitting at the heart of Quantum Computing so why not use this technology to model these complex quantum systems. And that's how QuantumBind was born.
What it does
QuantumBind is a hybrid quantum-classical pipeline that predicts the binding affinity of drug candidates for BACE1. We use this enzyme since this is the enzyme that can cause Alzheimer's so finding a molecule that can inhibit the effects of BACE1 would be groundbreaking to treating Alzheimer's.
The pipeline processes molecular data through four sophisticated stages:
1) Quantum VQE: We use the Variational Quantum Eigensolver (VQE) with a UCCSD ansatz to calculate the exact ground state energy of molecular fragments. This captures electron correlation that classical hardware simply cannot see.
2) Zero Noise Extrapolation (ZNE): To combat the "noise" of current quantum hardware, we run circuits at varying noise scales and extrapolate back to a theoretical "zero-noise" limit, ensuring high-fidelity data.
3) Structural Proteomics: The pipeline dynamically fetches the 3D BACE1 protein structure (UniProt P56817) from the AlphaFold database, extracting 10 critical geometric features of the binding pocket. AlphaFold was DeepMind's groundbreaking protein folding AI that accurately predicted the structures of 3D proteins.
4) Hybrid Neural Screener: A PyTorch neural network with an embedded Variational Quantum Circuit (VQC) layer fuses the quantum energy features with the AlphaFold structural data. The result of this neural network is a binding affinity score. If the affinity score is high (e.g. 0.9010), then the drug and the pocket of BACE1 are perfectly aligned making it a strong candidate for being an inhibitor to BACE1. Therefore, the drug is worth pursuing. But if the affinity score is low (e.g. 0.1020), then vice versa.
For the drug Verubecestat, QuantumBind returned a score of 0.88, identifying it as a high-potential candidate.
Finally, I put all this backend code and channel it through a Flask app and a frontend, consisting of HTML/CSS/JS, which is hosted on a .tech domain on MLH servers.
Additionally, I have a seperate file, hardware.py, that is capable of running my quantum algorithms on real IBM hardware. That means I can figure out exactly how well my algorithms work on a real quantum computer. This is also included in the Flask app.
How we built it
The quantum core of the project mainly consists of PennyLane for VQE construction and automatic molecular Hamiltonian generation. PennyLane is the standard for quantum chemistry since it has a dedicated library for qchem, making all of the tedious work handled under the hood allowing programmers, like me, to focus on the higher level logic.
While simulating this code with PennyLane, I also utilized IBM's Kingston 127-qubit superconducting processor through the Qiskit Runtime Estimator V2: (Job iD: d7didf15a5qc73dpt9f0)
Noise Mitigation: For constructing the ZNE algorithms, I made a custom-built implementation using PhaseDamping channels and a numpy polyfit extrapolation to create a linear relationship between the different noise values. This allows us to extrapolate back to zero, allowing us to estimate the ideal and noise free state of a quantum circuit.
For finding the binding affinity score, I use PyTorch neural network, integrated with a variational quantum layer, for end-to-end differentiability between classical and quantum layers.
After obtaining the structure from the AlphaFold API, I trained the model on 200 validated BACE1 measurements from the ChEMBL database.
Challenges we ran into
The absolutely biggest challenge I ran into was speed. Quantum computing programs naturally take a long time to process, due to the complex math behind these programs. That overall brought for a very bad user story since a researcher that would be working on a drug discovery project would have to wait a long time just to figure out if the drug is worth using or not. To solve this I had to use a multitude of methods such as threading, switching to more efficient methods and optimizing the computation time for the quantum circuits. Additionally, one of the original modules I planned to use, PySCF, lacked Windows support which led to difficulties in finding other workarounds to utilizing this popular and industry standard chemistry library. Finally, simulating a full drug, like Methylamine would require 30+ qubits and 38,000 Hamiltonian terms which is way outside the current capabilities of current free-tier quantum hardware. To develop an effective benchmark for my quantum algorithms, I used H2 while maintaining full protein analysis.
Accomplishments that we're proud of
Some things that I'm proud of include running live jobs on a 127-qubit IBM processor producing real world, accurate results that are verifiable on the IBM Quantum Platform. Since I verified that my algorithm works on real quantum hardware, I can scale this project in the future to make it a product that researchers and scientists can actually use to assist their research and speed up the drug discovery process.
Additionally, I'm proud of being able to bridge the gap between subatomic quantum physics and systems to macro-scale structural biology proving that end to end integration is possible for these kind of algorithms.
What we learned
I heavily advanced my skills in PennyLane and Quantum Computing in general. Before, I knew very little about quantum chemistry, but now I have left with a solid understanding of Hamiltonians, Jordan-Wigner transforms, VQE algorithms, etc. Additionally, this project can be expanded beyond BACE1 since it can create a universal Quantum Screening device for any kind of neurodegenerative diseases. I also learned that hybrid approaches (my quantum-classical PyTorch model) are viable and a powerful path forward for drug discovery today.
What's next for QuantumBind
Next, I would like to be able to split larger drugs into smaller fragments. Then I can run VQE on each of those fragments which will build a big library for quantum features. Then the next step, is to expand this Quantum Screening model beyond BACE1. I want to target other proteins, such as the ApoE4 which is a strong genetic risk factor for developing Alzheimers. I believe this proof-of-concept is a legitimate addition to the growing field of computational and quantum chemistry. I want to refine the findings from this hackathon to submit a real research paper and make a real impact in STEM.
Built With
- alphafold
- biopython
- chembl
- css
- flask
- github
- html
- ibm
- javascript
- matplotlib
- mlh
- numpy
- pennylane
- python
- pytorch
- qiskit
- render
- scikit-learn


Log in or sign up for Devpost to join the conversation.