-
The 4:3 version pf the Poster(Contains Introdution, Methodology, Results, Discussion and some performance plot)
-
The 3:2 ratio version of the Poster, content same as the first one
Title: Prediction of miRNA Diseases Associations Using Graph Convolutional Network Outline
Group Member Xuya Gao(xgao39), Zeming Liu(zliu185), Tongtong Zhao(tzhao34)
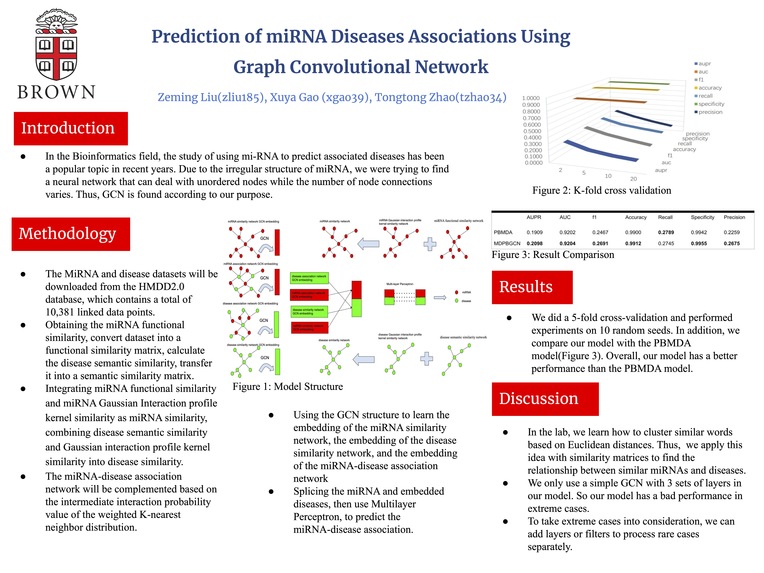
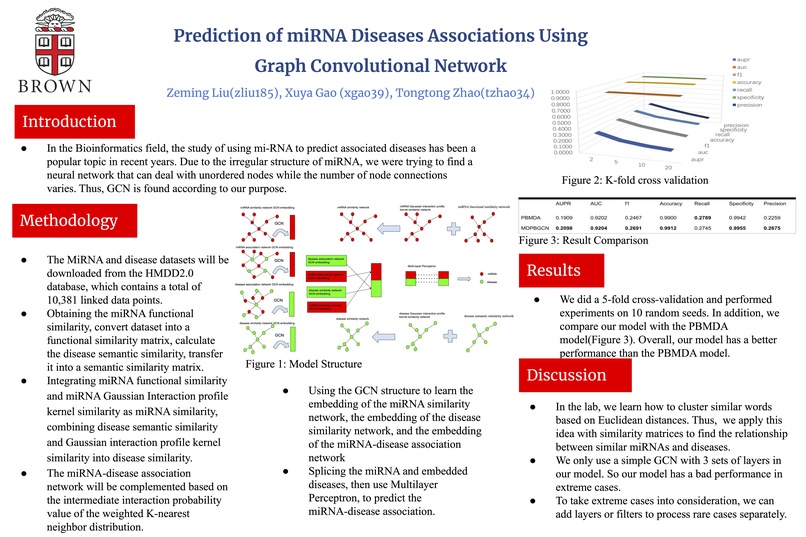
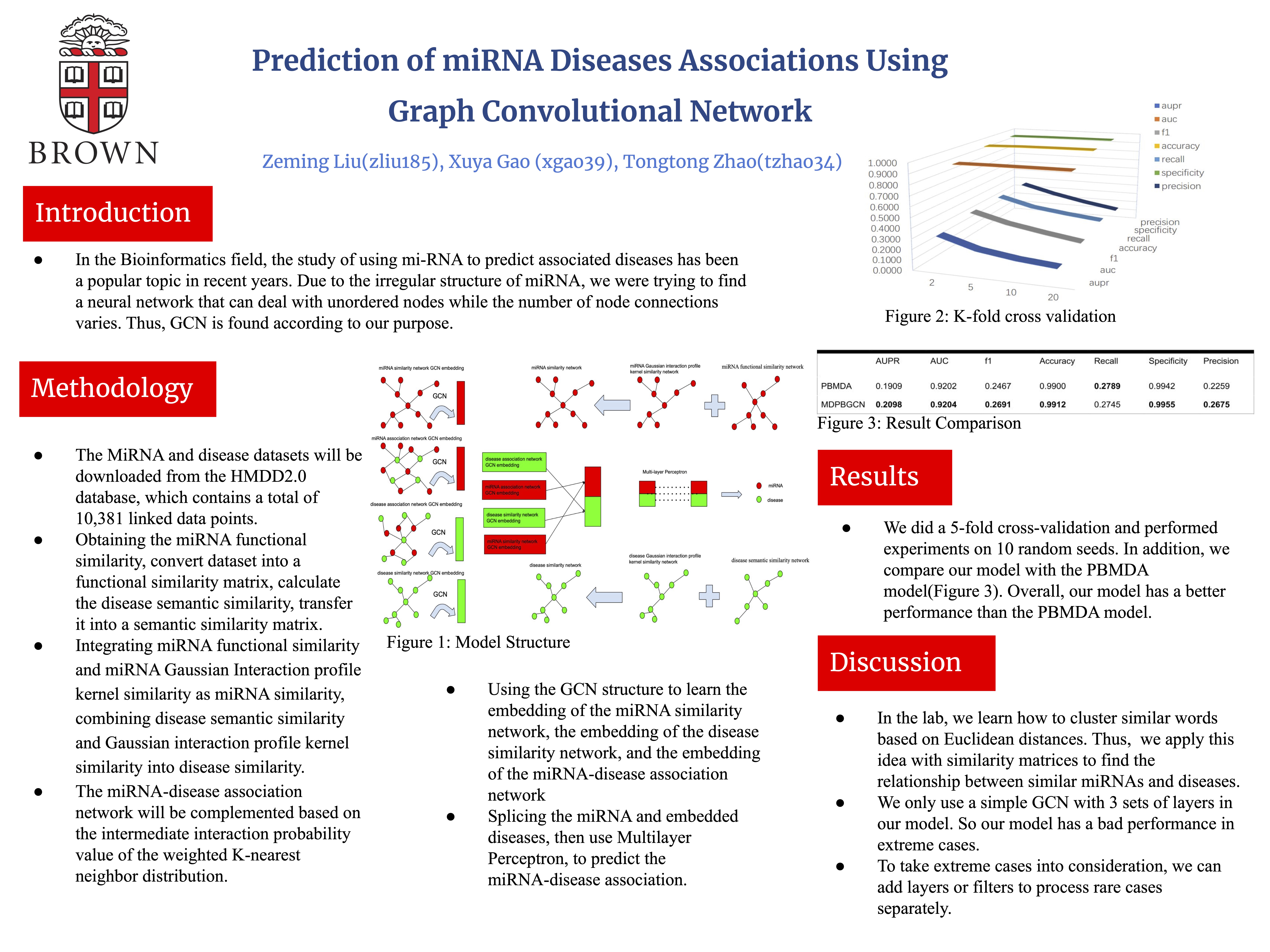
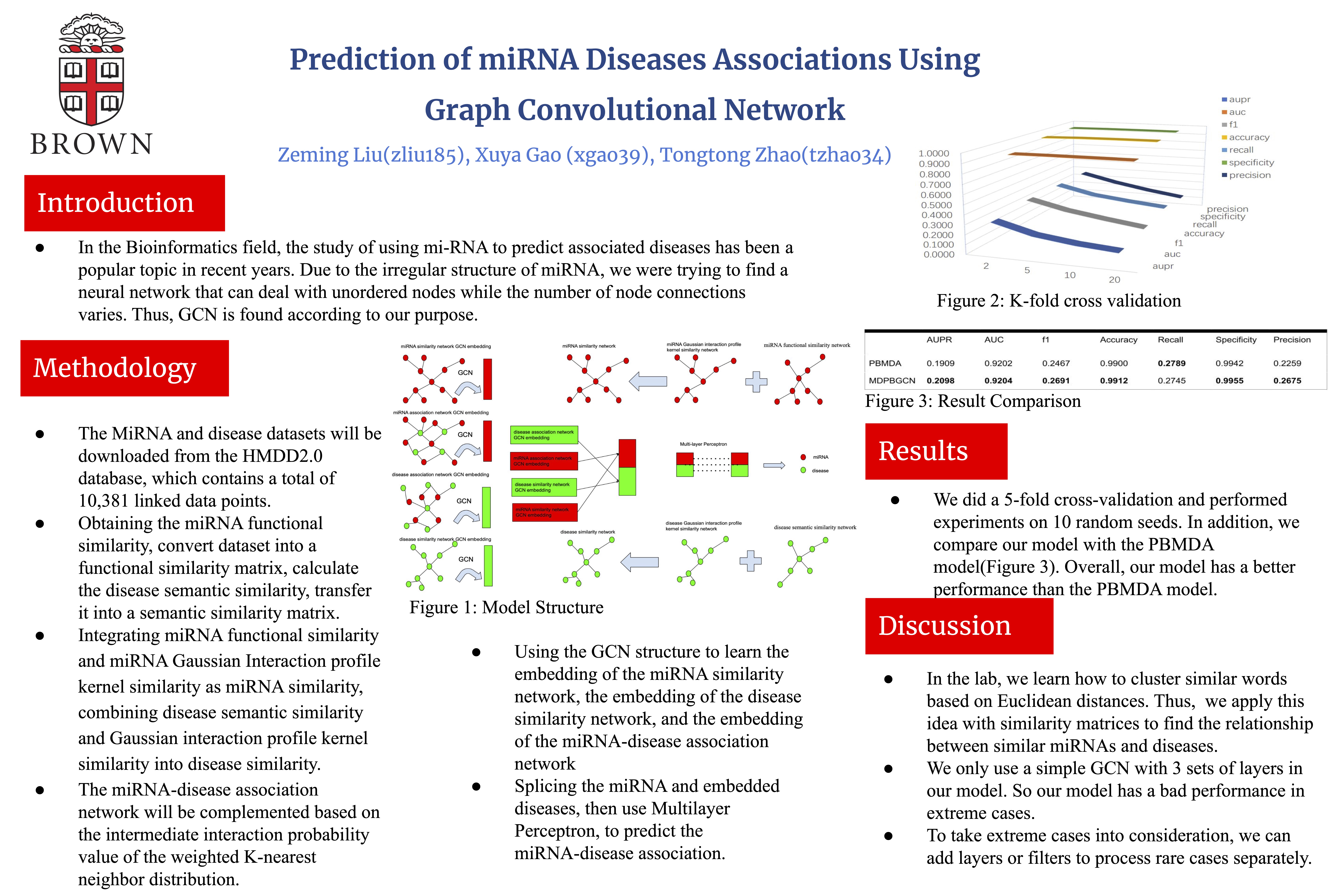
Introduction As we all know, mi-RNA is related to many diseases occurrences. In the Bioinformatics field, the study of using mi-RNA to predict associated diseases has been a popular topic in recent years. However, most researchers' techniques are models based on Convolutional Neural Network, which is hard to operate on unstructured data. Thus, we are motivated by finding a neural network that can deal with unordered nodes while the number of nodes connections varies; this neural network may define a good model, especially for mi-RNA. After reviewing a lot of papers, Graph Convolutional Network stands out from all other NNs, and it will be the one that helps us deal with irregular or non-structured data, which means it satisfies the characteristics we mentioned above. At this point, we believe that GCN will be helpful to generate a better model for the miRNA-associated disease prediction. Since our model is trying to predict diseases related to or caused by mi-RNA and compare to the real diseases, the problem type should be a structured prediction with binary classification.
Related Work The paper beyond our novel idea uses GCSENet, a model composed of GCN, CNN, and SENet, to predict the miRNA-associated diseases. The article mentions that GCSENet can detect the hidden relationships between miRNA and disease by three heterogeneous graphs to present an accurate prediction result. The model identified features by GCN from a heterogeneous graph has miRNAs, genes, and infections. Secondly, the author added a pre-set feature weight and feature inputs (combination of miRNA-gene and disease-gene associations) in CNN. Lastly, for SENet, the squeeze and excitation blocks were implemented to decide the feature channels’ importance and used the attention mechanism's means to improve valuable features. The author used the 10-fold cross-validation to assess the GCSENet’s performance. Their model’s AUROC score is 95.02%. Even we are not going to implement this paper, it’s better to have the paper’s weblink on file. The URL is: https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009048#abstract1
Data
The MiRNA and disease datasets will be downloaded from the HMDD2.0 database at http://www.cuilab.cn/hmdd. The HMDD2.0 database contains a total of 10,381 linked data points. Since those data were collected from different sources, some duplicate observations were in the database. hsa-let-7a-1 and hsa-let-7a-2 are miRNAs with the same mature sequence, but the names are slightly different because they are transcribed from different chromosomal DNA sequences. Thus they can be considered as the same species of miRNA. When we perform data preprocessing, they will be identified as hsa-let-7a uniformly. We will convert the associated data into matrices for storing, with rows representing miRNAs and columns representing diseases. Then we will remove the duplicate data of HMDD2.0, and 495 miRNAs, 383 diseases, and 5430 association data will be kept. MiRNA similarity data and disease semantic similarity data will calculate by cosine similarity, Euclidean distance formula, Jaccard similarity coefficient, and Gaussian interaction attribute kernel similarity. The miRNA-disease association network adjacency matrix, miRNA functional similarity matrix, and disease semantic similarity matrix will store in a CSV file in the form of matrices.
Methodology The model training process will use the random seeds to avoid better results due to randomness. The process will be repeated ten times to generate a total of 10 random seeds from 0 to 9 for data set dividing. For the division results of each seed, process k-fold cross-validation set k=5. The related miRNA-disease pairs are divided into five parts, and four miRNA-disease pairs are selected as positive samples of the training set each time. The exact number of miRNA-disease data are chosen as negative samples in the unrelated data(all unrelated miRNA-disease pairs are used as negative samples); this part includes all miRNA-disease pairs. Combine the positive and negative samples as the training set and the remaining one and all the negative samples as the test set for validating the model. The overall model is divided into five steps. The first step is to obtain the miRNA functional similarity based on the relevant database, convert it into a functional similarity matrix, calculate the disease semantic similarity, and convert it into a semantic similarity matrix. The second step will integrate miRNA functional similarity and miRNA Gaussian Interaction attribute kernel similarity as miRNA similarity, combining disease semantic similarity and Gaussian interaction attribute kernel similarity into disease similarity. The third step uses the WKNNP(a weighted k-neighborhood profile algorithm used to preprocess the interaction matrix) method to preprocess the data. In other words, The miRNA-disease association network will be complemented based on the intermediate interaction probability value of the weighted K-nearest neighbor distribution. The fourth step is to use the graph convolutional network structure to learn the embedding of the miRNA similarity network, the embedding of the disease similarity network, and the embedding of the miRNA-disease association network; The final step is to splice the miRNA and embedded diseases, then use Multilayer Perceptron, to predict the miRNA-disease association. Since we will implement a new model for the problem, the backup plan will not change the model or tune the parameters. We might simplify the steps that can not successfully give us the output, for example, streamline the third step using a more straightforward complementary method(still finding it).
Metrics First, to be considered a successful model, the lowest standard is to run the model without bugs and give a reasonable output. There are several known models for this prediction problem, so we will compare our model to the known models(maybe 4 known models). As we mentioned in the methodology part, the experiment will run ten times for each model to avoid the best result due to randomness. Since after the model training, we are going to compare the predicted diseases to the real diseases, which is considered a binary classification. Thus, we will use AUC, AUPR, F1, accuracy, recall, and precision to evaluate each model. Then, we are going to compare the metrics to assess our model. Moreover, our base goal is successfully run our model without bugs and generate a reasonable output, the target should be to close to at least one known model's result for the same topic prediction. The stretch goal is to have better performance in metrics than the known models.
Ethics The first ethical problem we are choosing is: Why is Deep Learning a good approach to this problem? Realistically, since we work on a model that takes mi-RNA and related data to predict diseases, we can use deep learning to build a good, highly reliable model to help patients prevent the diseases and save doctors time in determining the diseases. In the professional academic field, deep learning models are commonly used in lots of areas, and they generally have a better performance with reliable results than the other models. The reason why the deep learning model fits our problem is that the model will is capable to detect hidden, irregular complicated, non-linear connections from our original input, and keeping the pieces of information as time passes by. Since the pair-wise connection of miRNA amount is relatively large than we think, using the deep learning model will successfully solve the problem that the training set is too small, because every connection will be used as a training data point to train the model. The second question we are picking is: Who are the major “stakeholders” in this problem, and what are the consequences of mistakes made by your algorithm? The major stakeholders are doctors and medical researchers and patients, those people will benefit from the model directly. Another stakeholder will be the algorithm developer or software company(the company that turns the model/algorithm into a software or detection machine), they will get the benefit economically by selling the software or patents. If our algorithm makes a mistake, it might give them wrong suggestions during consultation and risk patients' lives. For the economic beneficiary, the consequence might be they get complaints and damage the company's reputation or even the future will be destroyed by the wrong algorithm since the algorithm is human life related.
Division of Labor Tongtong Zhao’s part will be data collection and integration of miRNA functional similarity and miRNA Gaussian Interaction attribute kernel similarity. Xuya Gaowill use WKNNPmethod to complement the miRNA-disease association network. Zeming Liu will use the GCN to learn the embedding of the miRNA similarity network, the embedding of the disease similarity network, and the embedding of the miRNA-disease association network using MLP to predict the miRNA-disease association. After all, all of us going to do the evaluation part together. Each of us will run the experiment on at least one model, the two persons with the shortest runtime will run the rest model to compute the evaluation metrics. The final report will be completed together with everyone covering the part they did.
References Li, Z., Jiang, K., Qin, S., Zhong, Y., & Elofsson, A. (n.d.). GCSENet: A GCN, CNN and Senet Ensemble Model for microrna-disease association prediction. PLOS Computational Biology. Retrieved April 12, 2022, from https://journals.plos.org/ploscompbiol/article?id=10.1371%2Fjournal.pcbi.1009048#abstract1
Peng, J., Hui, W., Li, Q., Chen, B., Hao, J., Jiang, Q., Shang, X., & Wei, Z. (2019, April 12). Learning-based framework for Mirna-Disease Association identification using Neural Networks. OUP Academic. Retrieved April 12, 2022, from https://academic.oup.com/bioinformatics/article/35/21/4364/5448859?login=true
Reflection
Introduction
As we all know, mi-RNA is related to many diseases occurrences. In the Bioinformatics field, the study of using mi-RNA to predict associated diseases has been a popular topic in recent years. However, most researchers' techniques are models based on Convolutional Neural Network, which is hard to operate on unstructured data. Thus, we are motivated by finding a neural network that can deal with unordered nodes while the number of nodes connections varies; this neural network may define a good model, especially for mi-RNA. After reviewing a lot of papers, Graph Convolutional Network stands out from all other NNs, and it will be the one that helps us deal with irregular or non-structured data, which means it satisfies the characteristics we mentioned above. At this point, we believe that GCN will be helpful to generate a better model for the miRNA-associated disease prediction. Since our model is trying to predict diseases related to or caused by mi-RNA and compare to the real diseases, the problem type should be a structured prediction with binary classification.
Challenges
The biggest challenge we encountered came from the problem of tuning the hyperparameters of the model, and the choice of algorithm for the similarity calculation. Since GCN has no application examples in this field, it was difficult to get a good choice of parameters from the reference papers. The hyperparameters that can be tuned include learning_rate, training epochs, the number of hidden layers, the number of neurons in the hidden layers, and keep_prob, a 1-dropout.
Besides, as our understanding of the dataset took us a considerable amount of time, we settled on the current model structure through intense discussions. Finally, we also did a lot of experimentation to ensure that the experiments were fair.
Insights
We did a 5-fold cross-validation and performed experiments on 10 sets of random seeds to obtain the experimental over in the table below. On 7 metrics, the model achieved AUPR 0.92, AUC 0.27, f1 score 0.21, accuracy 0.99, recall 0.27, specificity 0.996, precision 0.27.
We refer to the experimental results of PBMDA and our algorithm exceeds the AUC metric of the vast majority of algorithms.
Plan
In the final classification step, we simply applied a three-layer perceptron, and I think we could have further optimized our model with some more cutting-edge classification algorithms. Also in constructing the graph, we chose a weighted k-nearest neighbor complementary spectrum WKNNP, but I think we could improve the performance of the GCN with some similarity algorithms that would better preserve the structural information of the original data.
In addition, we only used generic data for our experiments, and now that miRNA and disease association data are emerging, I think we can do more experiments to validate our model and adjust the hyperparameters. We could also make comprehensive experiments across different state-of-the-art models.
Final Writeup
Group Member: Xuya Gao(xgao39), Zeming Liu(zliu185), Tongtong Zhao(tzhao34) Course Name: CS2470 Deep Learning
Introduction As we all know, mi-RNA is related to many disease occurrences. In the Bioinformatics field, the study of using mi-RNA to predict associated diseases has been a popular topic in recent years. However, most researchers' techniques are models based on Convolutional Neural Network, which is hard to operate on unstructured data. Thus, we are motivated by finding a neural network that can deal with unordered nodes while the number of node connections varies; this neural network may define a good model, especially for mi-RNA. After reviewing a lot of papers, Graph Convolutional Network stands out from all other NNs, and it will be the one that helps us deal with irregular or non-structured data, which means it satisfies the characteristics we mentioned above. At this point, we believe that GCN will be helpful to generate a better model for the miRNA-associated disease prediction. Since our model is trying to predict diseases related to or caused by mi-RNA and compare to the real diseases, the problem type should be a structured prediction with binary classification.
Methodology The model training process will use the random seeds to avoid better results due to randomness. The process will be repeated ten times to generate a total of 10 random seeds from 0 to 9 for data set dividing. For the division results of each seed, process k-fold cross-validation set k=5. The related miRNA-disease pairs are divided into five parts, and four miRNA-disease pairs are selected as positive samples of the training set each time. The exact number of miRNA-disease data are chosen as negative samples in the unrelated data(all unrelated miRNA-disease pairs are used as negative samples); this part includes all miRNA-disease pairs. Combine the positive and negative samples as the training set and the remaining one and all the negative samples as the test set for validating the model. The overall model is divided into five steps. The first step is to obtain the miRNA functional similarity based on the relevant database, convert it into a functional similarity matrix, calculate the disease semantic similarity, and convert it into a semantic similarity matrix. The second step will integrate miRNA functional similarity and miRNA Gaussian Interaction attribute kernel similarity as miRNA similarity, combining disease semantic similarity and Gaussian interaction attribute kernel similarity into disease similarity. The third step uses the WKNNP(a weighted k-neighborhood profile algorithm used to preprocess the interaction matrix) method to preprocess the data. In other words, The miRNA-disease association network will be complemented based on the intermediate interaction probability value of the weighted K-nearest neighbor distribution. The fourth step is to use the graph convolutional network structure to learn the embedding of the miRNA similarity network, the embedding of the disease similarity network, and the embedding of the miRNA-disease association network; The final step is to splice the miRNA and embedded diseases, then use Multilayer Perceptron, to predict the miRNA-disease association. Since we will implement a new model for the problem, the backup plan will not change the model or tune the parameters. We might simplify the steps that can not successfully give us the output, for example, streamline the third step using a more straightforward complementary method(still finding it).
Results We did a 5-fold cross-validation and performed experiments on 10 sets of random seeds to obtain the experimental over in the table below. On 7 metrics, the model achieved AUPR 0.92, AUC 0.27, f1 score 0.21, accuracy 0.99, recall 0.27, specificity 0.996, precision 0.27.
We refer to the experimental results of PBMDA and our algorithm exceeds the AUC metric of the vast majority of algorithms.
Challenges The biggest challenge we encountered came from the problem of tuning the hyperparameters of the model, and the choice of algorithm for the similarity calculation. Since GCN has no application examples in this field, it was difficult to get a good choice of parameters from the reference papers. The hyperparameters that can be tuned include learning_rate, training epochs, the number of hidden layers, the number of neurons in the hidden layers, and keep_prob, a 1-dropout.
Besides, as our understanding of the dataset took us a considerable amount of time, we settled on the current model structure through intense discussions. Finally, we also did a lot of experimentation to ensure that the experiments were fair.
Reflection
How do you feel your project ultimately turned out? How did you do relative to your base/target/stretch goals?
Our model performs much better than our expectations. In our base goal, we only expect our model to run without any bugs and generate a reasonable output. But our model can be run without any bugs and reaches good accuracy. As a result, our model performs better than the AUC metric of the vast majority of algorithms. This result satisfies our target goal and is close to our stretch goal.
*Did your model work out the way you expected it to? *
Our model works out the way we expected. It preprocesses inputs of miRNAs and diseases. Then our model generates a miRNA similarity network, disease similarity network, and miRNA-disease association network as we need. Later on, our model put those embedding networks into GCN and uses MLP to learn and predict miRNA-disease association. Our model follows our algorithm and generates the output which satisfies our requirements. In fact, our model performs better than our expectations and satisfies our target goal.
How did your approach change over time? What kind of pivots did you make, if any? Would you have done differently if you could do your project over again?
In fact, our team does not change our approach. We set up some reasonable goals at the beginning. Then we collect necessary information and study related materials based on our goals. We make up a plan for each person every week and use this plan to keep track of our progress. In this way, we can control our progress and adjust our plan if necessary. If we need to do this project again, we would like to spend more time studying the related materials. In this way, we can build a better model.
What do you think you can further improve on if you had more time?
First, we can read more papers and related materials about our topic. If we can read more papers, we might have better algorithms and structures in our model. In this way, we can improve our model’s performance. Second, we can collect more data. We use the HMDD2.0 database for miRNA and disease data. This database is good, but we can also collect other data to train our model. In this way, we can improve our model and reach a higher accuracy in the test.
*What are your biggest takeaways from this project/what did you learn? *
One of the most important things we learned in this project was teamwork. We set up our goal at the beginning, and we made a plan for everyone to keep track of our progress. We discussed problems with each other and provided anything we could to help each other. Also, we shared ideas with each other to improve our model and algorithm. In this way, we can finish the project and reach our goals. During this project, we learn that good teamwork will make 1 + 1 + 1 > 3. In the lab, we learn how to cluster similar words based on Euclidean distances. Thus, we apply this idea with similarity matrices to find the relationship between similar miRNAs and diseases.
Built With
- python
- tensorflow



Log in or sign up for Devpost to join the conversation.