-
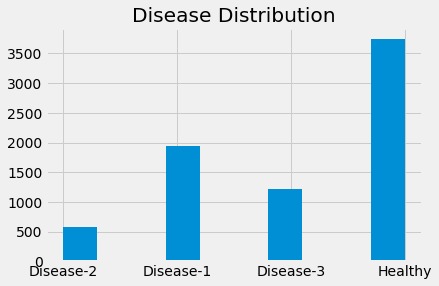
Distribution of Labels.
Using a Convolutional Neural Network to Perform Multiclass Classification of Disease in the Gut Microbiomes
Here, we use a convolutional neural network through the tensorflow library to perform the classification. We decided to use tensorflow because it is flexable, allowing for fine control of the model, and feature rich, meaning that we don't need to code everything from scratch. Additionally, we use many elements from the sklearn library for data analysis and preprocessing.
Data Analysis
Our first observation was that there were a large number of features that were pretty sparse. (Many of the features were zero.) Additionally, we knew that the values in each sample were not computed relative to eachother. For these two reasons alone, we need to clean this up before we can chuck it into a model. Taking a look at the frequency of each label, we found the following
| Label | Frequency |
|---|---|
| Healthy | 3741 |
| Disease-1 | 1949 |
| Disease-2 | 578 |
| Disease-3 | 1213 |
As we suspected, there was also not an even amount of data for each label. This is not a problem, as we can compute a weight for each lable to account for this difference in frequency when we train the model.
classes = list(np.unique(data['disease']))
class_weights = compute_class_weight('balanced', classes=classes, y=data['disease'])
class_weights = dict(enumerate(class_weights))
class_weights
{0: 0.9595946639302206,
1: 3.235726643598616,
2: 1.541838417147568,
3: 0.4999331729484095}
Data Preprocessing
Now that we have recorded the weights, we can work on transforming the data on our features into a more usable form. Because there are so many features and they are so sparse, this is a good situation to use dimensionality reduction. (It also helps us avoid the curse of dimensionality.)
We will first seperate all of the features into a dataframe and then normalize each sample. This makes each value the proportion of that type of bacteria detected by each test, a much more consistent and usable metric. Then, we will use Kernel Principal component analysis to condence the information into a smaller number of parameters (80) that we can use to train our model.
After cleaning our feature data, we can move on to our label data. We decided to encode each label using one hot encoding. This means that each label is assigned an index and each y value is converted to a vector that is all zeros except for the index of the label which is one. For example,
| Label | Encoding |
|---|---|
| Healthy | [1,0,0,0] |
| Disease-1 | [0,1,0,0] |
| Disease-2 | [0,0,1,0] |
| Disease-3 | [0,0,0,1] |
We could assign each label an integer or float value, but this approach seems more natural for this problem, and it can be generalized to a multilabel classification problem if a patient happens to be unfortunate enough to be suffering from multiple diseases at once.
# Encode the Y values using OneHot encoding.
Y = np.zeros(shape=(len(Y), len(classes)))
for i in range(len(Y)):
Y[i,classes.index(data['disease'][i])] = 1.0
Validation Data
Before we begin to train our model, we must save some data that we won't train on in order to test how accurate our model is after training. We will use a random 30% of our data to test the accuracy of our model. We will calculate the F1-Score and Cohen's Kappa after we are done training.
Model Construction
Now we construct the model. We will use a feed forward neural network where every other layer is a dropout layer. This means that in each training batch, 5% of the edges in these layers will be deleted and the weights of the remaining edges will be boosted a small amount to compensate. This is an extremely effective method to avoid overfitting the data, which is of great concern to us because we have a relatively small amount of data relative to the number of features (before reduction.)
Because we have categorical data that is not well balanced, we decided to use SigmoidFocalCrossEntropy as our loss function which handles this data well (https://arxiv.org/pdf/1708.02002.pdf). Because we are using cross-entropy as the loss function, we must use the sigmoid or softmax function as our activation function, which is not a problem. We decided to use sigmoid except for the output layer where we decided to use softmax. This is because softmax forces the sum of the outputs to be one, allowing us to interperit the output as a probability distribution.
# Build the model. (We need to use softmax because we have cross entropy as the loss function.)
# The dropout layers are there to prevent overfitting with a lot of training on our dataset.
dropout_rate = 0.05
model = Sequential([
Dense(400, activation='sigmoid'),
Dropout(dropout_rate),
Dense(100, activation='sigmoid'),
Dropout(dropout_rate),
Dense(20, activation='sigmoid'),
Dropout(dropout_rate),
Dense(4, activation='softmax')
])
Validation Metric Results
Finally, we comput the F1-score and Cohen's Kappa for the validation to assess how good our model is.
Y_prediction = model.predict(X_validation)
for idx, label in enumerate(classes):
prediction = [np.argmax(arr) == idx for arr in Y_prediction]
actuial = [np.argmax(arr) == idx for arr in Y_validation]
print(f"{label} F-Score:\t", f1_score(prediction, actuial))
prediction = [np.argmax(arr) for arr in Y_prediction]
actuial = [np.argmax(arr) for arr in Y_validation]
print("Cohen's Kappa:\t\t", cohen_kappa_score(prediction, actuial))
Disease-1 F-Score: 0.7170818505338078
Disease-2 F-Score: 0.2633228840125392
Disease-3 F-Score: 0.6765899864682001
Healthy F-Score: 0.7998266897746966
Cohen's Kappa: 0.5647816371716303
Discussion
The F-Scores look pretty good, except for Disease-2. This is likely due to the fact that there was significantly less data available for this label than for the other labels. More data collection would definately help with respect to that label specifically. Moreover, if there was data collected from patients experiencing multiple diseases would enhance the model performance because it would allow us to assume that each Disease is independent from the other diseases. This is not in fact the case, but it is much closer to the truth than assuming that each disease percludes the posibility of having any other disease.
Built With
- python
- sklearn
- tensorflow

Log in or sign up for Devpost to join the conversation.