-
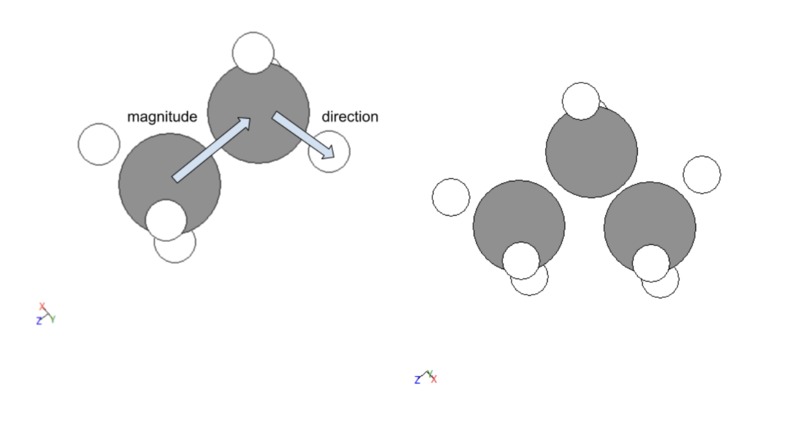
Figure 1
-
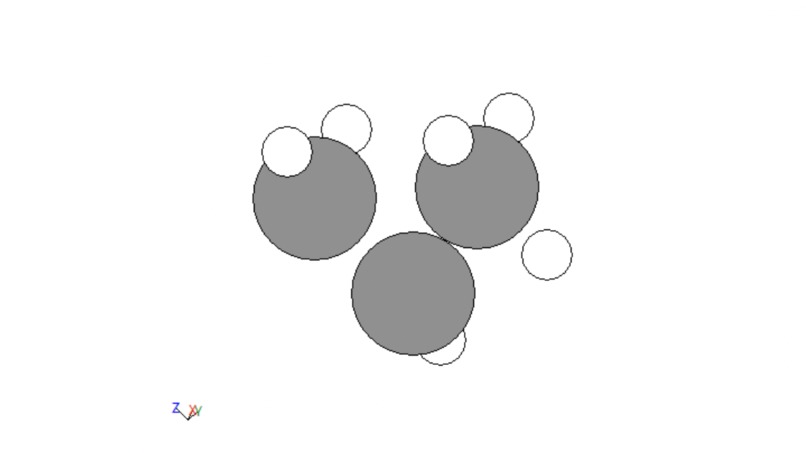
Figure 2
-
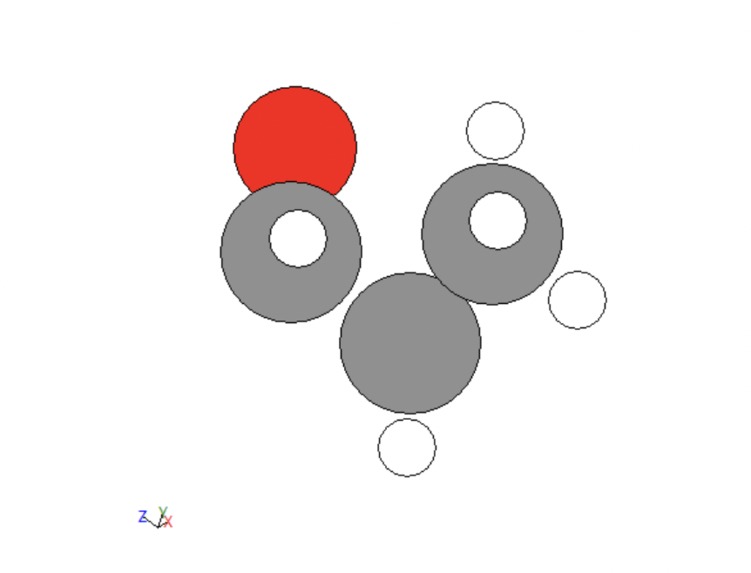
Figure 3
-
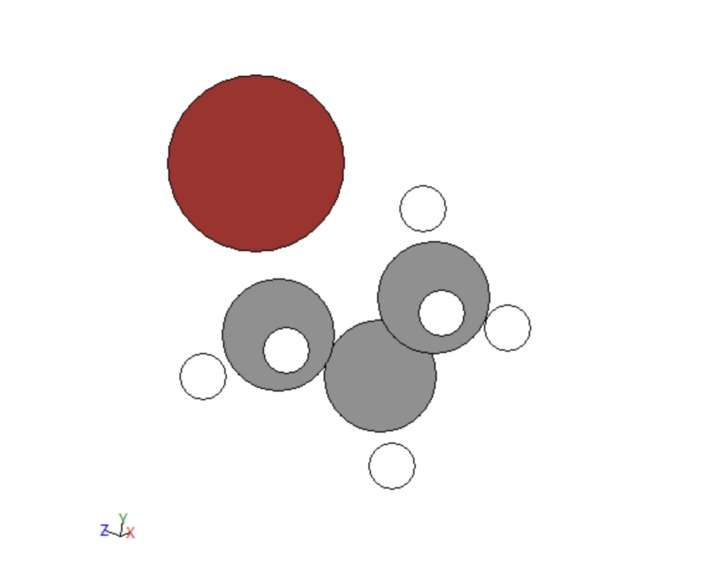
Figure 4
-
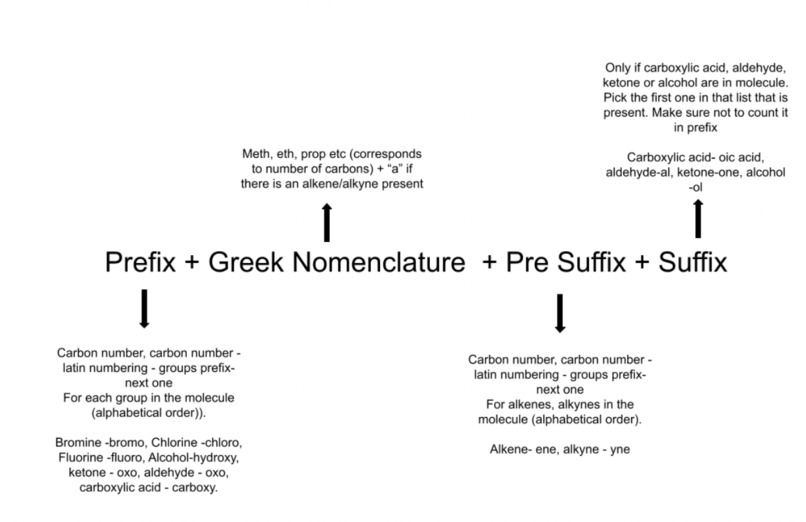
Figure 5
-
Title: OrgoNet: 3D Modeling of Complex Organic Compounds Using a DNN
Who:
Niko Bhatia - nbhatia
Ugo Piovan - upiovan
Manuel Lopez- manuellopez2
Cameron Fiore - cfiore
Introduction:
The motivation for this topic is to speed up the slow process of visually representing molecule structures in Python’s ASE extension. Currently, there are very limited molecules (<50) in the library, so it is more a demo than a useful tool. To create new molecule structures, it guesses initial molecule locations and takes 8+ hours to slowly adjust to the final equilibrium position using intermolecular forces. We believe that a DLNN can be trained to take in the molecule name, output the distances between atoms, and quickly output the 3D molecule structure with Python’s ASE extension.
Our problem can be classified as a Regression problem. We are working with labeled data, and we map the molecule name to a continuous vector representing the 3D positions of all atoms in the molecule.
Translating the InChI: adapting neural machine translation to predict IUPAC names from a chemical identifier: https://jcheminf.biomedcentral.com/articles/10.1186/s13321-021-00535-x
The article above discusses the development of a neural machine translation model to convert the International Chemical Identifier (InChI) into International Union of Pure and Applied Chemistry (IUPAC) names, which is a standardized naming system for chemical compounds. The authors explain how they trained their model using a dataset of over 50,000 compounds and achieved high accuracy rates in predicting IUPAC names. Towards the end, they also discuss potential applications for this technology, including improving chemical database searches and aiding in the discovery of new compounds.
Struct2IUPAC -- Transformer-Based Artificial Neural Network for the Conversion Between Chemical Notations: https://www.researchgate.net/publication/347140196_Struct2IUPAC_--_Transformer-Based_Artificial_Neural_Network_for_the_Conversion_Between_Chemical_Notations
Data:
We have developed our own set of data for this project. Given that there wasn’t a repository of IUPAC names and structures large enough to train an ML model, we had to devise a methodology to make our own. Ultimately, 440,000 molecules were created, named and split into 40,000 for testing and 400,000 for training.
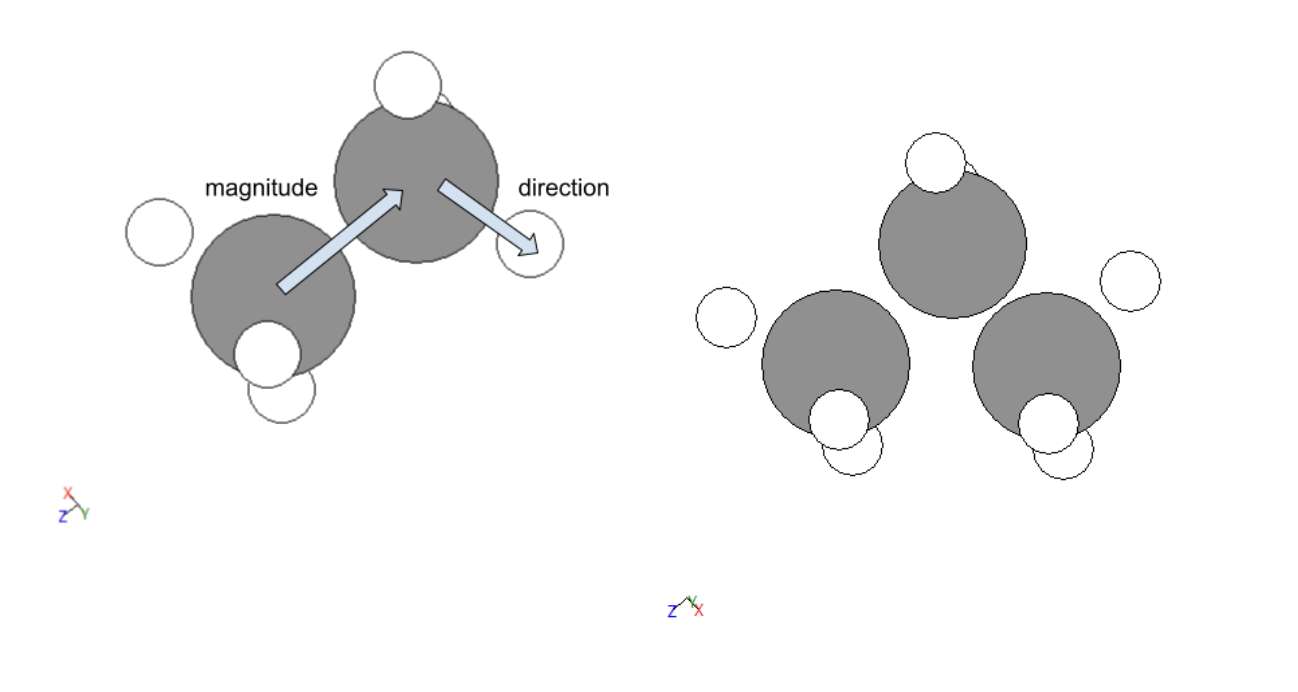
The methodology for creating data was split into five portions, the first was creating a molecule without functional groups (composed of just a line of carbons with the external ones having three hydrogens attached and the internal ones having just two). This was done by importing methane or ethane from the atomic simulation environment’s data. For molecules with more carbons than ethane vector operations were utilized to place a carbon in the corresponding space, the three hydrogens were then shifted to be on the new external carbon and two new hydrogens were added to the former external carbon with identical relative positions to the other internal carbon. An illustration of this is given below. This process was abstracted for larger carbon chains.
Figure 1: Vector process for creating molecules
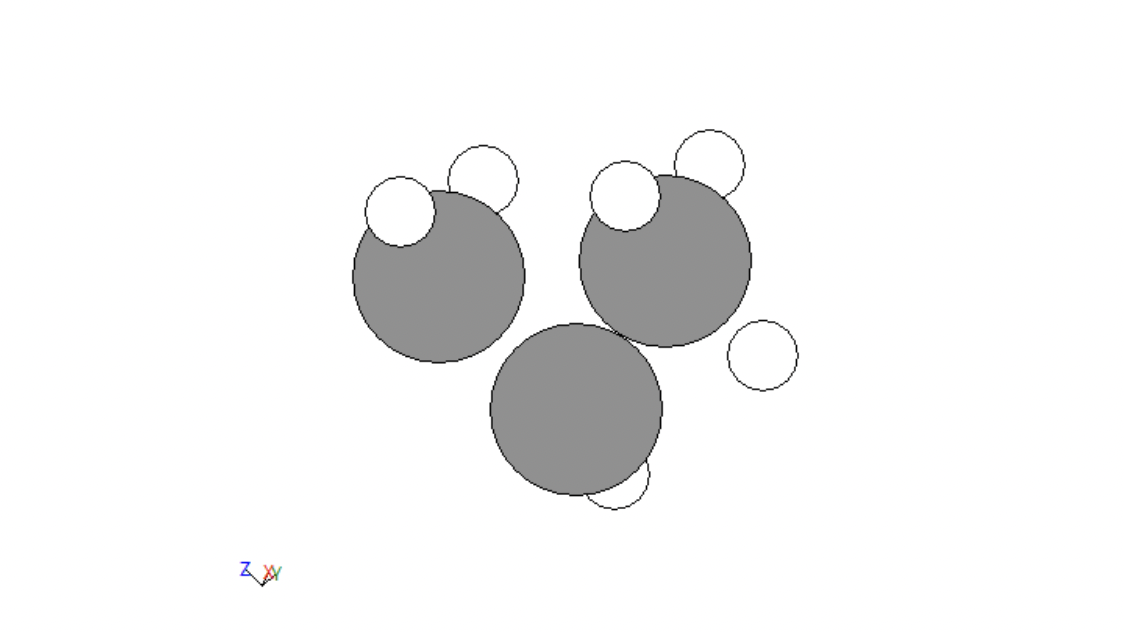
The next portion of data development was adding functional groups. The following groups were allotted: alcohols, bromines, chlorines, fluorines , alkenes, alkynes, aldehydes, carboxylic acids, and ketones. Functions were created to handle each of these. For the simple cases of bromines, chlorines, and fluorines, a hydrogen was fed into each function, changed to be the desired atom, and then the distance to its carbon was adjusted. For alcohol this same process was done, a carbon was changed to an oxygen and the distance to its carbon was adjusted but then a hydrogen was added to the alcohol on the same axis that linked it to the hydrogen at the distance between the original hydrogen and its carbon. Alkenes and alkynes were handled by removing two or four hydrogens from two neighboring carbons. For ketones two hydrogens on the same carbon would be fed into the function, one would be removed and the other would be converted to an oxygen. For aldehydes this same thing would be done but on an external carbon. Finally, for carboxylic acid the same process would be repeated but then the third external hydrogen would be fed into the alcohol function. A few examples are given below.
Figure 2: Alkene
Figure 3: Carboxylic acid
Figure 4: Bromine
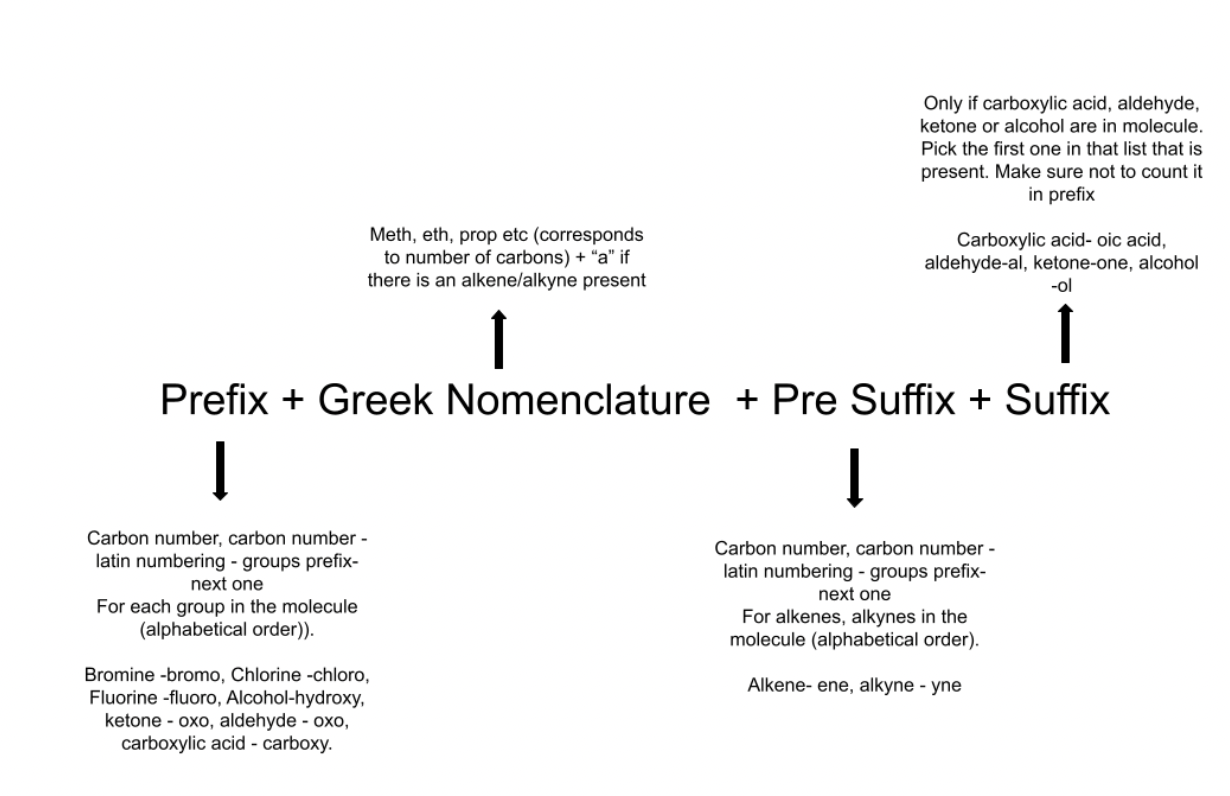
The third step to developing data was to create a significant number of molecules with random functional groups in random positions. Although this portion became somewhat convoluted, a simple explanation is that a loop was created for carbon chains ranging from size 1 (methane) to size 10 (decane). The version without functional groups then had its number of atoms, atomic positions, element types, and name (more to come) printed to a text file. Arrays were made to keep track of the number of hydrogens that hadn’t been converted to a functional group, the types of functional groups that were possible for each hydrogen (for example carboxylic acids are only possible on external hydrogens, alkenes are only possible when a hydrogen is on one carbon and on an adjacent carbon) and which functional groups were on each hydrogen (needed for naming). For each carbon size a random functional group was selected from a list of possible groups, then a random hydrogen was selected, the functional group was added to the molecule and its information was added to the output text files. Given that the range of sizes, groups, and hydrogens was random, a large number of molecules could be tested. As such, for the training data this loop was run through 10,000 times and for the test data this was run through 1,000 times. The final portion of this was developing a name for each molecule. As was stated previously, when each molecule is developed a simple array accompanies it that describes which functional groups are present. The array has a length equal to the number of hydrogen atoms on a molecule (two times the number of carbons plus two). The numbers in the array correspond to a functional group. A diagram of the IUPAC naming rules is given below. The code breaks the process into four functions. Each requires only the array described above. The first finds the Greek numbering system for the carbon chain. The number of carbons is converted to “meth”, “eth”, “prop” etc. as necessary, it is also noted if there is an alkene or alkyne group, if not then an “a'' is appended to the prefix.
The next step is the pre suffix, this part is only necessary if there is an alkene\alkyne present, otherwise only “an” is returned. If there are alkenes or alkynes they are counted, numbered and returned with each number representing a position followed by a latin counting system followed by “yn” or “en” depending on which group it is, for example “1-2-dien”. Next is the suffix, only the functional group of the highest priority is affected by this, it applies for only carboxylic acids, aldehydes, ketones, or alcohols in that order for priority. If there is one present it will be removed from the functional groups list and the suffix will be given as “oic acid”, “al”, “one”, or “ol” depending on which type it is, if none of these are present the suffix will be “e”. Finally the prefix is taken by removing the group given to the suffix, then using the same latin prefix and dash system as for alkenes the prefix is found by listing these alphabetically (for example 1-bromo-2-3-dichloro). The name is given by adding the strings found in this order: prefix + greek + pre suffix + suffix.
Figure 5: Nomenclature


Log in or sign up for Devpost to join the conversation.