-
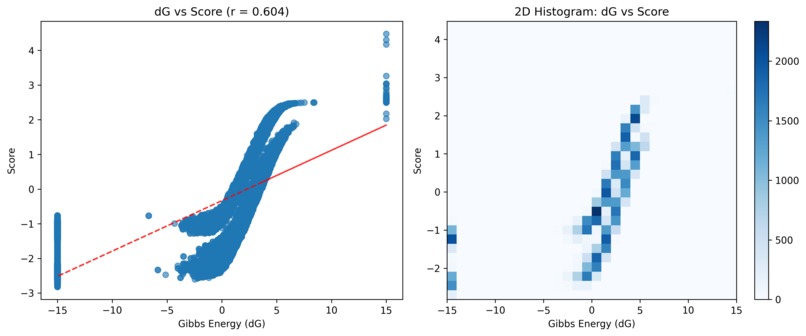
Comparison with Delta Gibbs
-
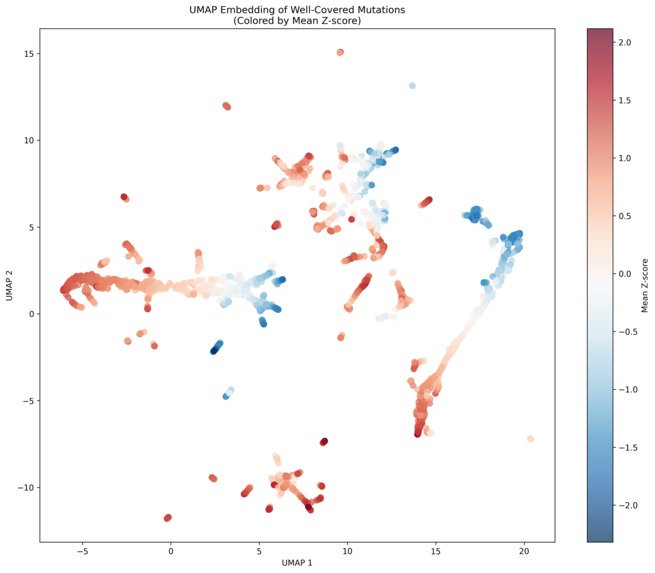
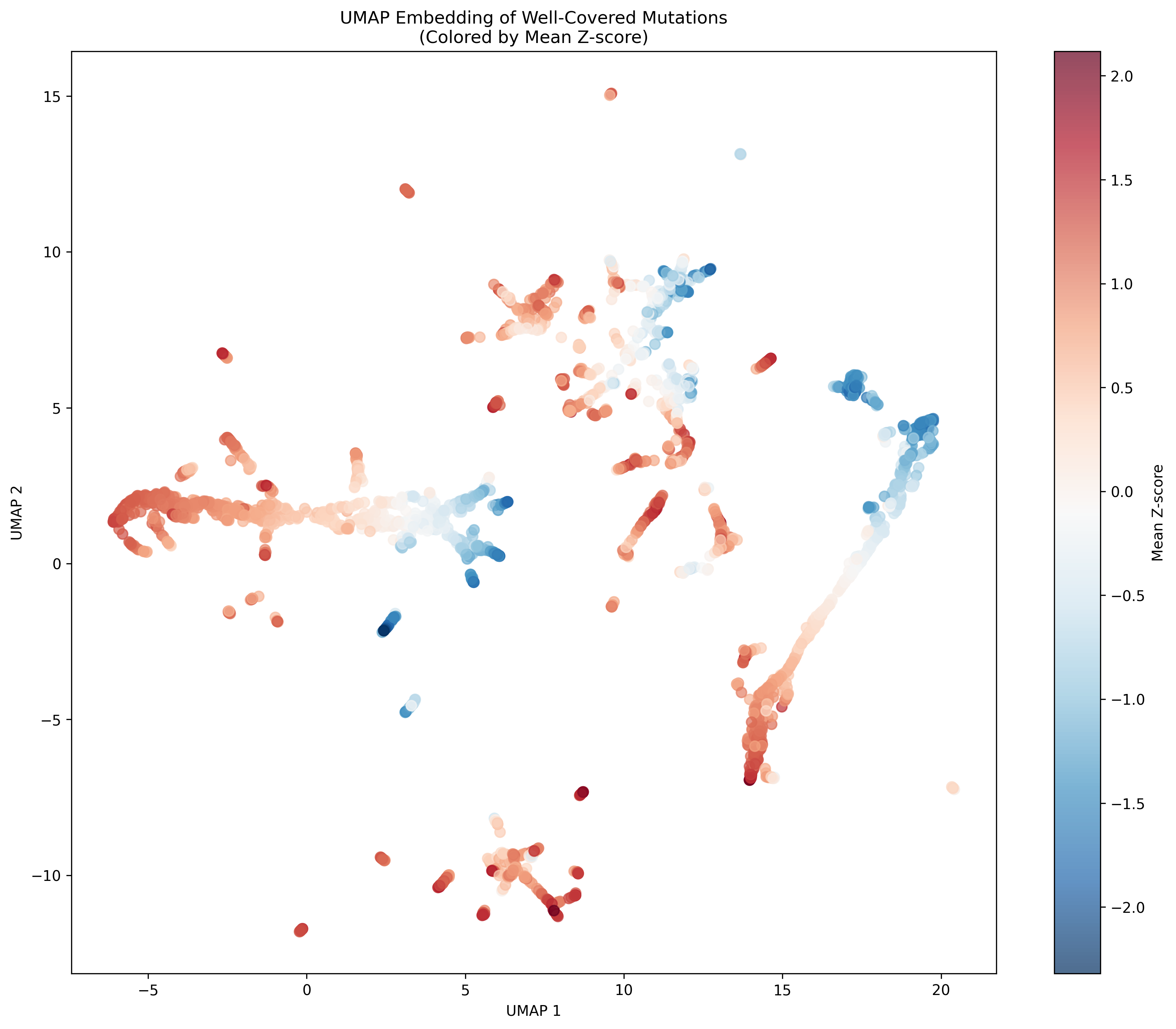
Clusters
-
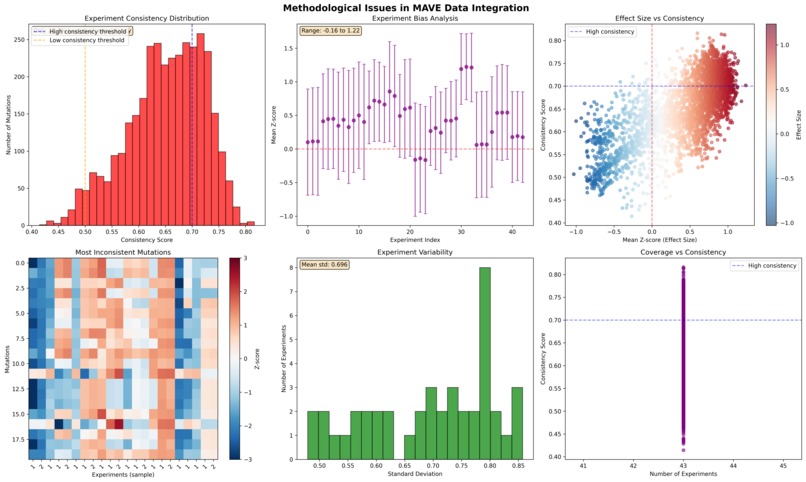
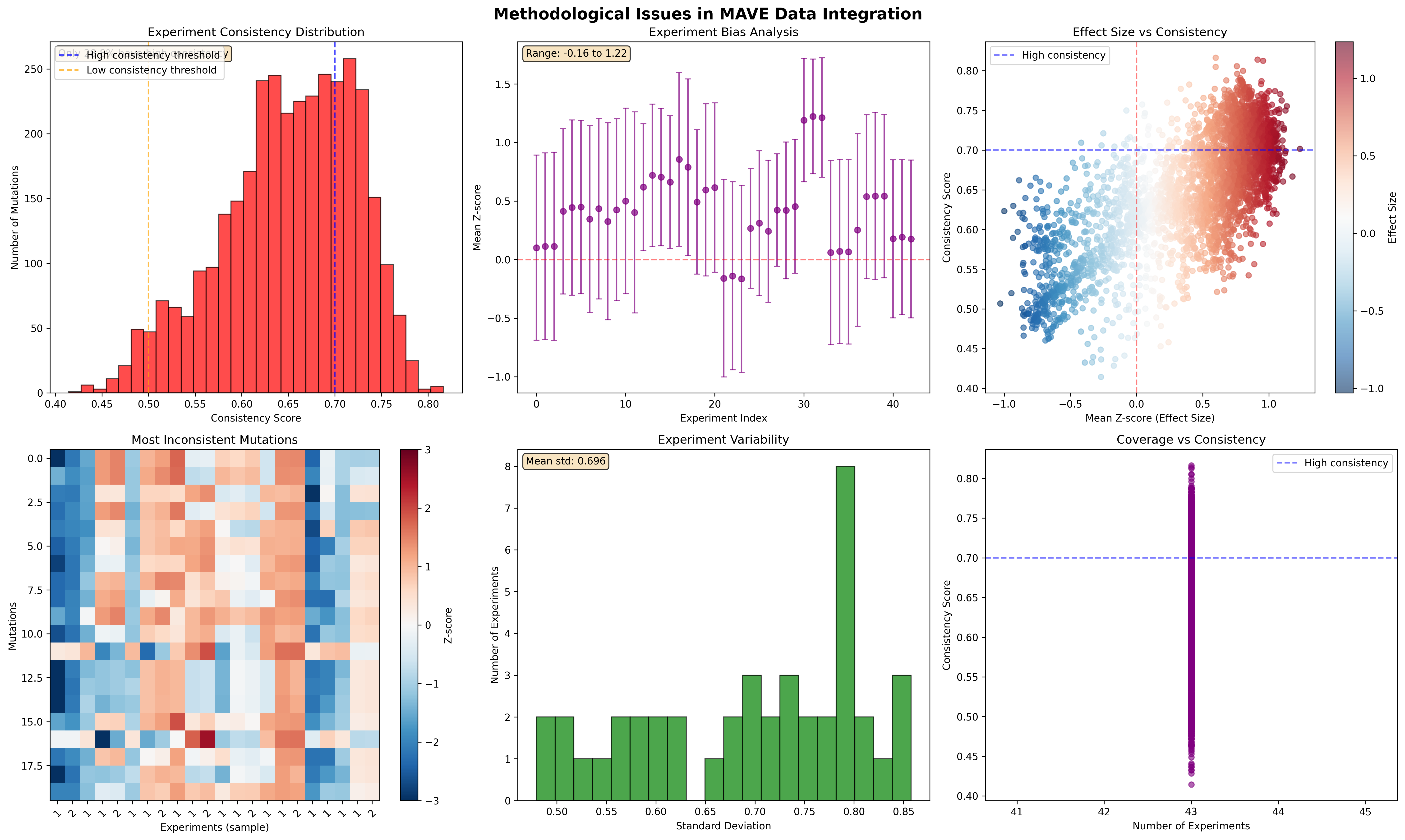
Z Score Analysis
Inspiration
Using ML Skills in Bio
What it does
Multiplexed assay of variant effect (MAVE) data integration across experiments presents significant methodological challenges that limit clinical utility. We present a comprehensive framework for analyzing cross-experiment consistency, implementing quality control protocols, and developing imputation strategies for sparse MAVE datasets. Using SPTAN1 variant data as a case study, we demonstrate that only 60.2% of mutations exhibit high consistency across experiments (consistency score ≥ 0.7), while experiment-specific biases range from -0.083 to 1.055 z-score units. Our analysis reveals fundamental limitations in current integration approaches and proposes generative models (Dirichlet and Boltzmann LDA) as promising alternatives to traditional imputation methods. The framework establishes quality control metrics and validation protocols essential for reliable clinical interpretation of MAVE data.
How we built it
Cursor carried hard
Challenges we ran into
Our original proposal was a lot more ambitious, but after looking through the data and thinking about potential methods, we picked something basic as a baseline/starting point towards this normalization.
Accomplishments that we're proud of
What we learned
Plan better
What's next for Normalizing SPTAN1 Across Different Experiment
Use a different method than Z scores and consider using generative models (Dirichlet/Boltzman LDA)
Built With
- mavedb
- python

Log in or sign up for Devpost to join the conversation.