Inspiration
Inspired by applied data science and the brilliant scientists who traditionally develop drugs through labor-intensive methods, we set out to accelerate the process.
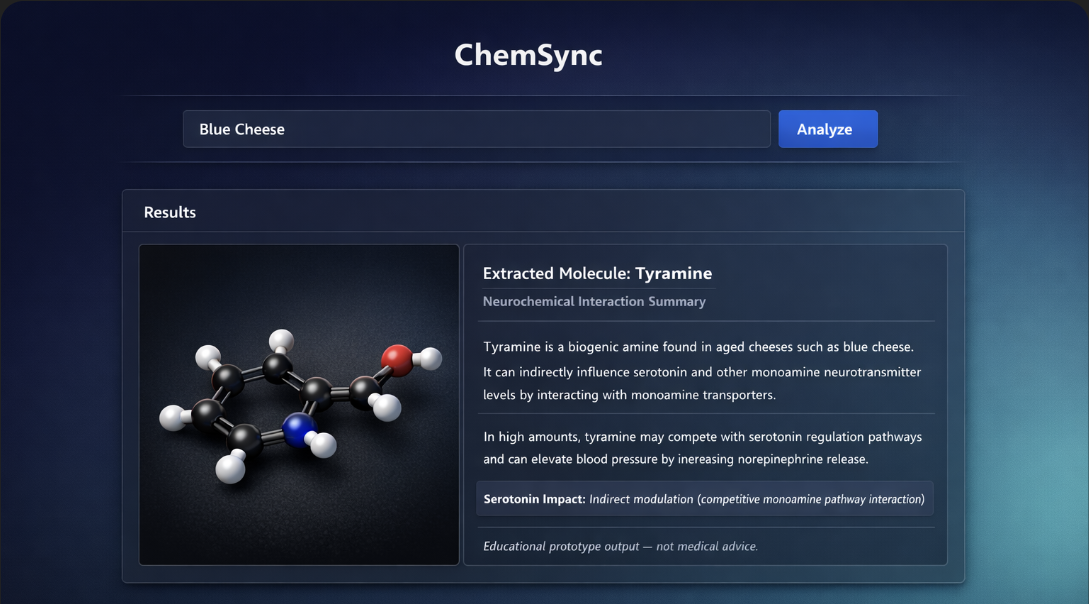
What it does
Our system allows users to input a substance (food, drug, or chemical compound) and estimate its potential psycho activity by predicting binding affinity to selected serotonin receptors. We focused on key serotonin receptor subtypes that are central to antipsychotic medications and emerging depression treatments.
How we built it
We used RDKit in Python to convert molecular structures into graph representations, where atoms are nodes and bonds are edges. A graph neural network (GNN) was trained on a large receptor affinity dataset to predict how strongly a compound binds to specific serotonin receptors.
On the frontend, we integrated an LLM to interpret user input and identify valid chemical substances before passing them into the prediction pipeline.
Challenges we ran into
Predicting biological effects beyond receptor binding proved challenging, as real-world pharmacology is complex and multi-factorial. Our largest prediction errors were approximately 5× the average value. While this may seem high, such variability is common in experimental lab assays and remains within realistic biological ranges
Accomplishments that we're proud of
Our graph neural network successfully captured structural features such as molecular shape, binding sites, and binding affinities. Notably, it was able to approximate pharmacophore-like patterns — a process that traditionally requires significant time and domain expertise.
What we learned
We gained hands-on experience applying graph neural networks to real-world biochemical data. We also learned the challenges of working with noisy experimental datasets and translating scientific problems into machine learning frameworks.
What's next for Neurotransmitter Binding Affinity Prediction Using GNN
Future improvements include expanding the model to additional receptor types, decomposing molecules into functional moieties for more granular learning, and increasing training data diversity to generate more refined pharmacophore predictions.

Log in or sign up for Devpost to join the conversation.