-
-
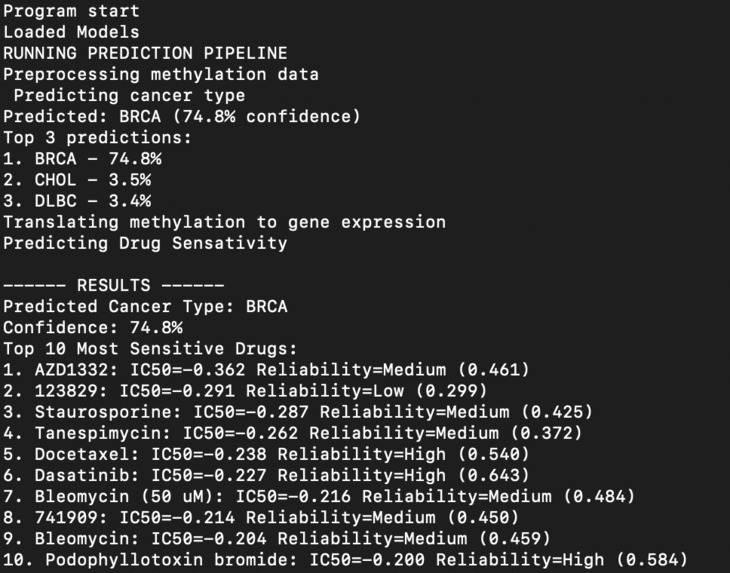
Sample Results
-
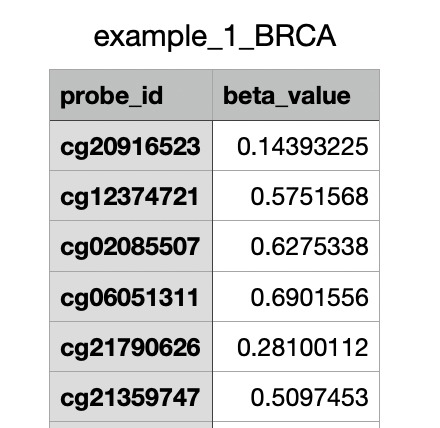
Example portion of dataset
-
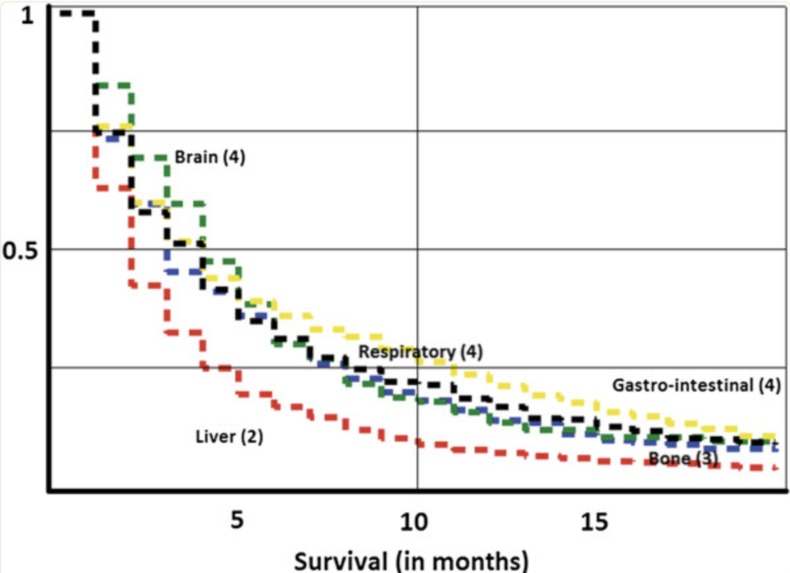
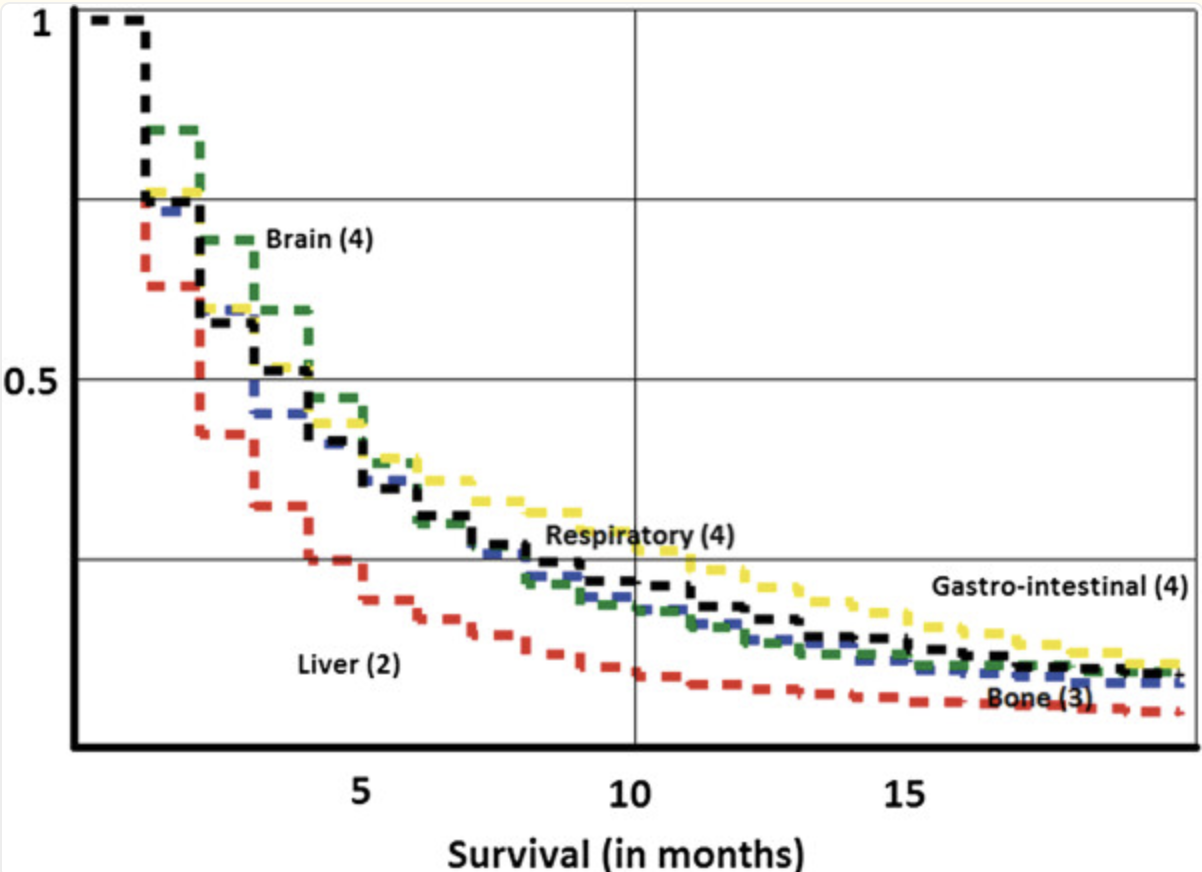
Mortality rate for CUP
-

Part of the code

Inspiration
Cancer of Unknown Primary Site (CUP) accounts for around 3-5% of cancer cases, and has a median patient survival time of only 3 months (1). This makes rapid diagnosis and treatment incredibly important to increase patient survival rates. Current options for treatment are often invasive, slow, or costly making it inaccessible to many patients (2). It was critical to build a cheaper alternative to help patients with CUP. Our product takes a DNA methylation profile, which can be done for < $300 (3), and outputs a cancer prediction type and a ranked drug sensitivity profile for the patient.
The most recent public research and initiatives by private companies are resulting in accurate methylation tests that can be completed in only a few days, and as little as several hours (4). Various technologies, like Automated Cartridge-based Cancer Early Screening System (ACCESS), can perform methylation testing with minimal resources and at mobile or remote testing sites (5).
What it does
We use a three stage deep learning pipeline that performs cancer type classification and drug sensitivity prediction from the methylation profiles. The system first uses a residual MLP network to predict one of 33 cancer types. Then, a secondary encoder decoder model is used to turn the original methylation profile and turn it into a gene expression vector. The last model predicts drug sensitivity (IC50 values) to each type of cancer, providing an immediate and cost effective way to predict cancer types and immediately assess drug effectiveness. Additionally, the model produces a confidence score for each of the 270 possible drugs. This allows doctors to make informed tradeoffs between potentially more effective drugs and higher confidence in effectiveness.
How we built it
First, we used a TCGA Pan-Cancer methylation with over 450k data points which we treat as the primary input system. We filtered the raw beta values and removed patients with high amounts of missing values. We also had TCGA RNA-seq which was used as a training target for the translation model which we used to do gene overlap with GDSC expression data. Then we found a GDSC dataset that provided drug sensitivity with IC50 values across 270 drugs.
Using 3 deep learning models, we used PyTorch and trained on a Macbook Pro to process the many gigabytes of data. On our first model which predicted the cancer type, we achieved over 94% accuracy. For our translator model, we achieved a median pearson r of 0.66. Then for the IC50 predictor, we achieved a median pearson r value of 0.53. All 3 of our models achieved high accuracy and the data is outputted along with confidence values to allow for educated decisions.
Challenges we ran into
Downloading the datasets and aligning the data types was very tricky. We used 2 dataset systems where one was used to predict the drug sensitivity and the other predicted the type, but had different data formats. Additionally, as we are not bioinformatics or biology majors, it was a struggle to understand how to process the datasets and generate a way to convert the methylation profiles into the gene expression vector that is compatible in a way for the drug sensitivity dataset that we used.
Accomplishments that we're proud of
We are proud of successfully processing and combining 2 separate datasets that use different data formats and we managed to extract insights using both. Additionally, training 3 separate deep learning models that use this and having them all produce high accuracy in a short time frame.
What we learned
We learned a lot about bioinformatics and gene expression data during the creation of our project. Prior to this, we had very little experience working with genomic data, primarily with sequence alignment, so we learned a lot about how to create a model to translate the methylation profile into gene expressions. We also learned a lot about how to assess drug sensitivity and different metrics that are used in the real world.
What's next for Methylation Profiled Cancer Identification and Drugability
We would like to translate our models over to nanopore sequencing for both the drug sensitivity and the CUP identification. Nanopore sequencing is typically cheaper and more modern than the current methylation datasets that we used, and translating over could provide added insights and accuracy to our models. Additionally we would like to create more effectively edge deployed versions of our pipeline that would allow our models to run on low powered devices (like a mobile phone) so that they can be used in areas without extensive infrastructure in order to provide equitable and actionable cancer diagnosis.
Sources
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3565900/
- https://www.cancer.gov/types/unknown-primary/patient/unknown-primary-treatment-pdq
- https://www.cgm.northwestern.edu/cores/nuseq/services/next-generation-sequencing/dna-methyl-seq.html
- https://www.hopkinsmedicine.org/news/articles/2024/04/the-story-of-epigenetics
- https://pmc.ncbi.nlm.nih.gov/articles/PMC12013524/

Log in or sign up for Devpost to join the conversation.