-
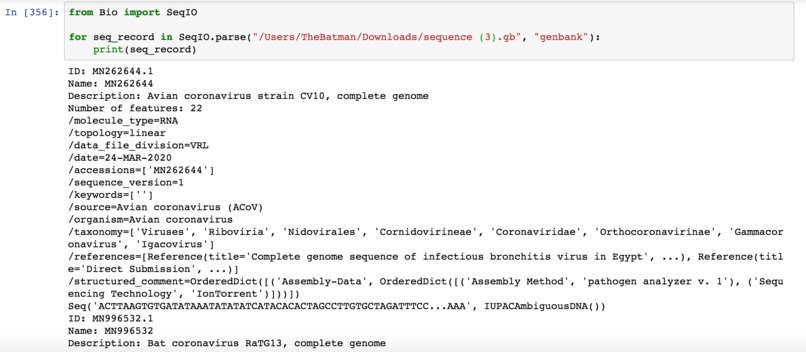
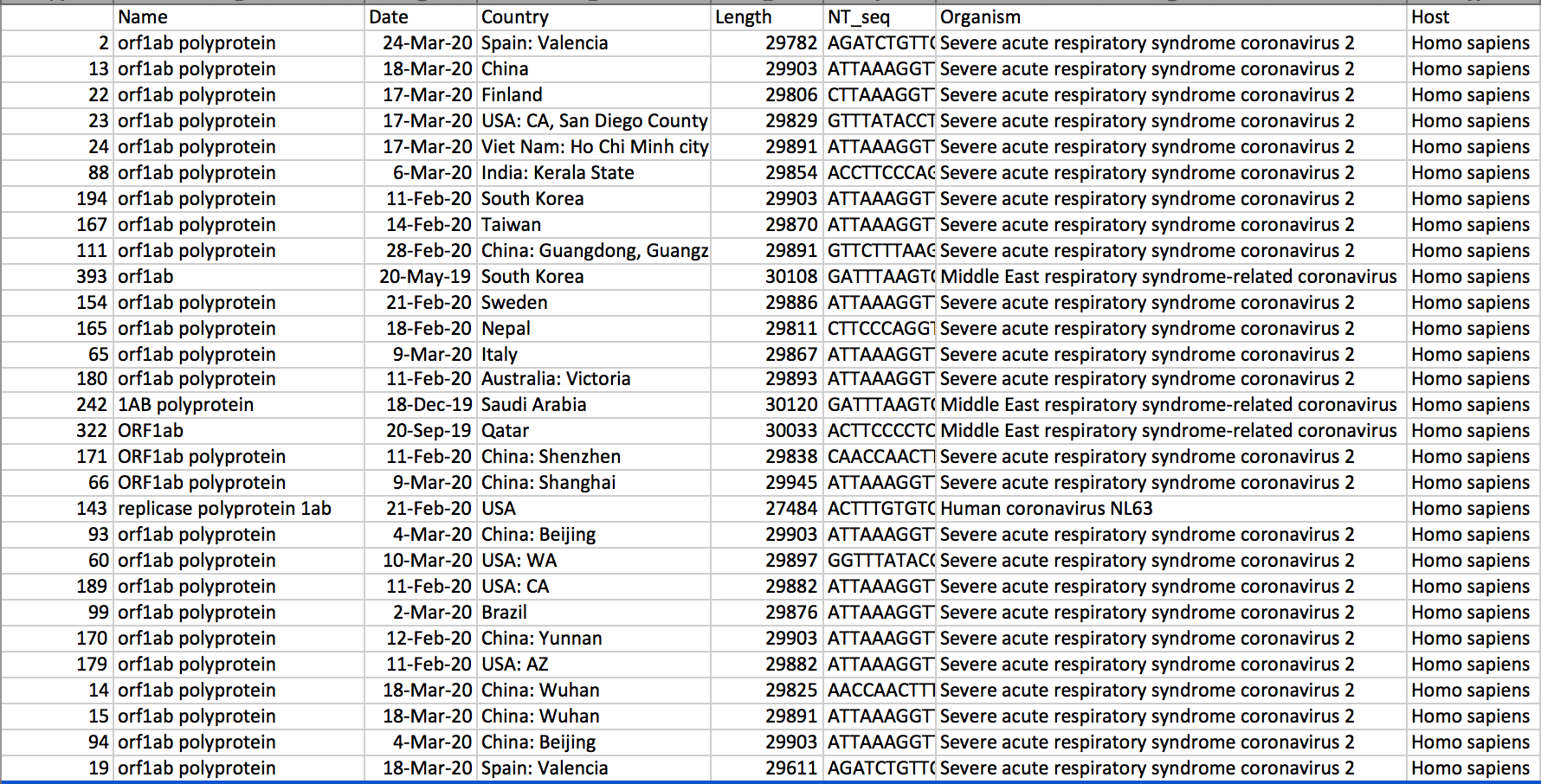
The Biopython library was used to parse GENBANK files downloaded from NCBI. Appropriate information was extracted from the raw data fields.
-
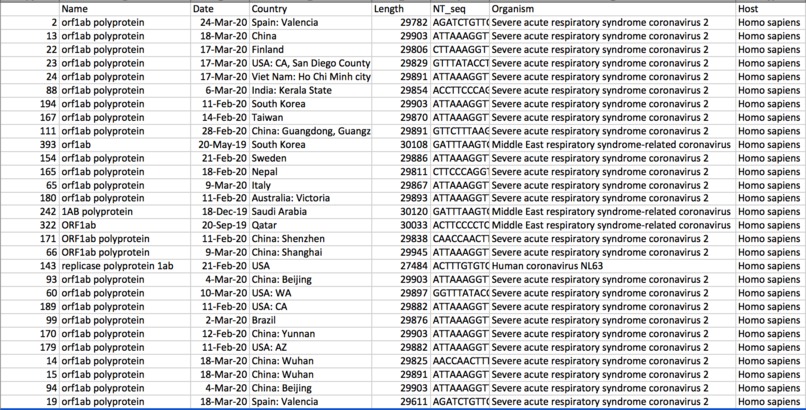
A CSV file was then created after pulling relevant genomic and geographic data. It organized locations, sequence lengths, hosts, and dates.
-
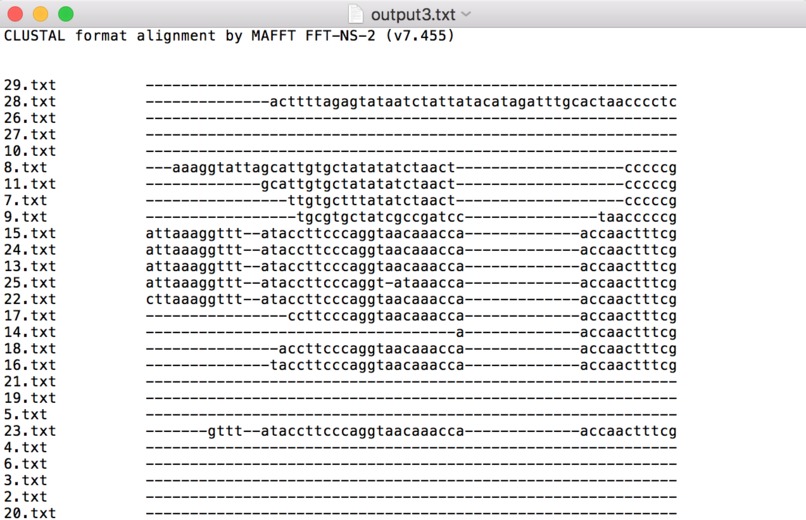
A screenshot taken from the MAFFT bioinformatics program that automates the alignment of sequences using multiple sequence alignment.

Inspiration
It's hard to ignore how quickly COVID-19 has spread around the world. In a matter of weeks, it's raced from Wuhan to Europe to the United States. Given its incubation period and the globalization of the world, tracking the movement of the virus has proven difficult for scientists and epidemiologists. However, as cases have popped up, over 400 genome sequences have already been uploaded to NGDC (National Genomics Data Center of China) as well as other online databases. Using this information, then, what if there were some way to correlate corona's movement with its genome?
How I approached it
All viruses evolve and mutate rapidly. Their large population sizes and fast lifecycles make it very likely that their genomes differ in different cases. Therefore, if more mutations exist between two sequences of the virus compared to another, perhaps that might be an indicator to the virus's movement. Intuitively, we can then track the spread of a virus through the evolution of its genome.
I looked on NCBI's website (National Center for Biotechnology Information), which holds a myriad of public genomic information. I specified search criteria for genomic DNA for coronaviruses that were at least 20,000 nucleotides long (to approximate the entire genome of the virus), getting about 3000 results. From this, I narrowed it further to sequences that were taken from Homo sapiens as hosts, ignoring those taken from pigs, bats, and other animals, ending up with about 400.
I then applied multiple sequence alignment using the bioinformatics program MAFFT. Essentially, this reveals similarities and differences between different sequences, and it's commonly used to look into evolutionary relationships. In addition, I extracted geographic locations and dates from website data and looked at possible relationships between all three variables.
Challenges I ran into
Something I immediately underestimated was the amount of time it took to conduct multiple sequence alignment. I originally wanted to look at sequence differences in humans AND animals, potentially to examine how the virus had evolved once it took on human hosts. Running the program on about 600 of these sequences took forever, and, in the time that I'm writing this, still has not been completed. I instead had to compromise and took about 30 sequences instead, making sure to select from a wide variety of countries and dates to ensure there was no specific bias in the data.
A second challenge I ran into was quantifying and visualizing these differences in genomes. Specifically, the output of MAFFT only shows locations in the genome that differ or are similar (see the screenshot attached). Converting this to a readily understandable format, such as rankings from most similar to most different, was a challenge, while converting it further to a state that can elucidate geographic location was something that wasn't able to be finished due to time constraints. However, I definitely believe that given a visualization that pinpoints a virus on the map as well as highlighting how different it is from the original Wuhan strain, this would clarify the message of this project much more.
Insights
To put it briefly, all sequences shared stretches of very similar nucleotides, which makes sense because they're all coronavirus genomes. However, substantial mutations seemed to occur in many of the strands. Upon examining their location and date, sequences that exhibited more mutations/differences from the other strands seemed to be taken from February and before, whereas the other sequences that shared more similarities were taken from roughly similar time periods in March. However, there are definitely exceptions to this trend, as some recent March sequences were found to have as many differences as the sequences from February and before.
In attempting to extract geographic insights, no immediate and obvious connection was found. While China sequences seemed to be more similar compared to ones from Italy and California, for example, no concrete correlation could be pinpointed due to the seemingly random scatter of the data. However, I am dealing with only 30 data samples, so if the originally planned 600 could be used, I could have more of a basis to make conclusions on.
Accomplishments that I'm proud of
I'm excited by computational biology, but it's been something that I never have had an opportunity to get hands-on experience with besides coursework. Having the opportunity to delve into data by myself and manually work with bioinformatics tools without a professor-written guide was really cool, and I'm proud that I was successful in my goals. Specifically, I learned a lot more genome alignment from working with MAFFT, virology from reading about evolution of viruses, and COVID-19 itself.
Built With
- mafft
- python
Log in or sign up for Devpost to join the conversation.