-
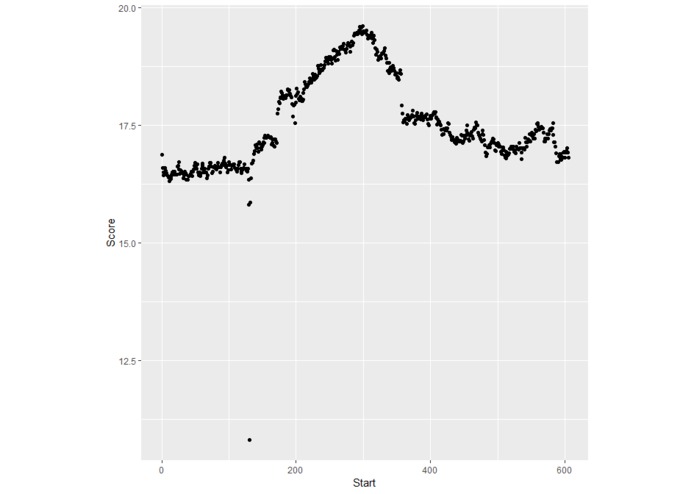
Plot created by distance data values created by ICARUS.
Inspiration
Proteins make up the human body and allow us to function. Unfortunately, these proteins also have the capability of causing great harm when they do not perform correctly, playing a major factor in diseases of modern society. To combat this and improve the physical welfare of modern society, we must better understand proteins through their unique characteristics. Proteins carry a special structure-function relationship, where the structure of proteins allow for the variety of activities they can do.
However, thousands of proteins remain uncharacterized in terms of both structure and function. This leaves humans in a vulnerable state, as we may not be able to understand diseases well due to not being able to link illnesses to a specific protein.
To combat this, machine learning algorithms have been developed that analyze amino acid sequences to determine potential structure features. A problem with multiple current programs is the use of greedy algorithms, which may not yield the best possible fold recognition results. Researchers of DNA sequences have recently published work recommending the use of the mean-shift algorithm in substitute of current programs, and have obtained improved results. We take this approach to protein sequences in order to better understand domains/folds (characteristic protein features) in an attempt to improve current protein knowledge.
What it does
This program transforms mean-shift clustering into a classification algorithm. Sequences are first cleaned and duplicates are removed in order to develop better data groups during mean-shift clustering. These sequences are then converted into vectors that can be used by machine learning. Mean shift clusters proteins based on their similarity and provides cluster center coordinates. The program determines a radius that represent the average distance between points associated with a cluster centroid and the cluster centroid. This radius is used to create a circle. This circle can be used to predict whether or not sequences contain a certain fold, depending on whether or not coordinates are inside or outside of the circle. ICURAS converts an input sequence to coordinates and determines the distance between its points and the closest cluster center. If the distance is less than or equal to the circle's radius, the program predicts the sequence to carry a specific fold. ICURAS takes a rolling window approach and tests multiple sub-sequences within your sequence input if possible, and will provide distance values, associated sequences, and a threshold score for prediction.
How we built it
ICURAS was built using scikit-learn's API. Sequences were derived from the Pfam database. Purge from the MEME Suite was used in conjunction in order to process fold protein sequences. R's ggplot2 was used in order to plot data from this project. Code was written in jupyter notebook with Python 3.7.
Challenges we ran into
Before TreeHacks, neither of us knew how to use machine learning algorithms. However, discussion with TreeHack mentors improved our understanding and allowed for the project to continue. Data pre-processing was a new concept, and required literature review in order to understand a reasonable way to convert 20 amino acid letters into a numerical form.
Evaluation metrics of whether or not sequences contained a certain fold were also unclear at the beginning of the project, but figured out after data visualization.
Accomplishments that we're proud of
UHRF1 is a multi-domain protein that is recognized to be overexpressed in several forms of cancer. These domains differ in structure, and vary in size greatly (60 to 200 residues). Using ICURAS, we used UHRF1's domain as test set and were able to identify all 5 domains in sequence.
What we learned
Machine learning, machine learning, and machine learning! How coding is not set in stone with how its methods should be used was definitely a highlight, considering our conversion of a clustering algorithm to a classification system.
What's next for ICURAS - Machine Learning Protein Fold Recognition
If we are able to gain access to cloud computing, we are interested in further developing cluster centers for the current set of 650,000 protein domains. We would use this knowledge in order to develop a thorough public resource, where researchers can input sequences of little known knowledge and potentially receive leads on how to learn more about these uncharacterized proteins.
How domains act with one another, and whether domains often appear with one another would also be of interest in a machine learning situation.
Collaboration with protein modeling groups/programs would also be of interest, as ICURAS would be able to provide structure-independent assistance to current programs which rely on information like residue distances in order to determine structure. Predicting certain folds within a sequence may provide protein modeling a template as aid.
References: James, B. T., Luczak, B. B. & Girgis, H. Z. MeShClust : an intelligent tool for clustering DNA sequences. 46, 1–10 (2018). Bailey, T. L. et al. MEME S UITE : tools for motif discovery and searching. 37, 202–208 (2009). Asgari, E. & Mofrad, M. R. K. Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics. 1–15 (2015). doi:10.1371/journal.pone.0141287 Darosa, P. A. & Harrison, J. S. A bifunctional role for the UHRF1 UBL domain in the control of hemi-methylated DNA-dependent histone ubiquitylation. Mol. Cell (2018). Cerami, E. et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2, 401 LP-404 (2012). J Yang, R Yan, A Roy, D Xu, J Poisson, Y Zhang. The I-TASSER Suite: Protein structure and function prediction. Nature Methods, 12: 7-8 (2015). A Roy, A Kucukural, Y Zhang. I-TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols, 5: 725-738 (2010) Y Zhang. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics, vol 9, 40 (2008). The Pfam protein families database in 2019: S. El-Gebali, J. Mistry, A. Bateman, S.R. Eddy, A. Luciani, S.C. Potter, M. Qureshi, L.J. Richardson, G.A. Salazar, A. Smart, E.L.L. Sonnhammer, L. Hirsh, L. Paladin, D. Piovesan, S.C.E. Tosatto, R.D. Finn. Nucleic Acids Research (2019) doi: 10.1093/nar/gky995

Log in or sign up for Devpost to join the conversation.