HIV-1 DeepTropism
Inspiration
HIV-1 is still a pandemic with around 37 million infected globally. The past decades showed a great improvement in the development of drugs that could help patients fight the infection and increase life spam. More than 25 drugs of 6 different classes are available for treatment. One of the drug classes called Chemokine receptor antagonists targets cell co-receptors, CCR5 or CXCR4, used by the virus to infect human cells. Maraviroc is an approved Chemokine receptor antagonists and blocks the interaction of the virus with CCR5 present on the patients cell membrane. As most of the early infection is caused by CCR5 tropic viruses, Maraviroc can be used with success to treat the patient. As the infection progresses the virus can evolve to use CXCR4 to infect new cells, and Maraviroc becomes inefficient. So before prescribing Maraviroc for patients, it is important to know which virus tropism (R5-tropic or X4-tropic) is causing the infection. Different machine learning methods were applied during the past decades to predict HIV tropism, but there is still room for improvement, as the performance of the tools available is still deficient.
What it does
DeepTropism is a Deep Neural Network for defining HIV-1 tropism based on the V3 protein loop sequence.
Building the Dataset
In order to develop a model that could show a good performance against the broad diversity of HIV-1, we have organized a dataset based on 5 previous published datasets on viral tropism (. The total number of sequences was 9550 and length from 21 to 35 amino acids. After labeling each sample as CCR5 tropic (coded as 0) or CXCR4 tropic (coded as 1) we removed the duplicated sequences to avoid bias, resulting on a final dataset of 3608 unique sequences. This dataset was used for training, validation, and testing. Dataset unique sequences :
- CCR5: 2783 samples (labeled as 0)
- R5X4: 485 samples (labeled as 1)
- CXCR4: 340 samples (labeled as 1)
Challenges I ran into
Feature extraction
One of the main challenges about dealing with genetic data is how to transform the information we have as a string into arrays and matrices to be used as input for training. In order to compare each protein sequence we first aligned the dataset with unique sequences using Muscle Aligner with gap open value of -15. The resulted fasta file was formed of a 60 position alignment of all the sequences. These aligned sequences were used as raw data for the next step. To convert the protein sequence into tensors we decided to used a simple approach of one-hot encoding of each amino acid on the sequences. A traditional amino acid one-letter code table has 20 different aminoacids, an X representing any molecule, and a few representing dubious amino acids (B, Z, and J), totaling 26 positions. For each letter on our V3 loop sequence, we created a numpy array of zeroes of size 1 by 26, and replaced the corresponding position of the aminoacid with a 1, as each amino acid was represented by one position in the array. So each aligned protein sequence of 60 characters resulted in a numpy array of 60 by 26. This matrix was then linearized into an array of 1 by 1560 and used as the input tensor of our Deep Neural Network.
Defining the architecture of our Neural Network
Of othe main questions Data Scientist ask when faced with a new problem related to Deep Learning is which architecture to use. As always a good advice is to start simple and gradualy improve complexity. That was exactally what we did. We decided to used a simple DNN architecture of 3 fully connected layers of 1560, 250 and 100 layers. The output layer was formed by the 2 nodes representing our binary outcome (Figure 2 attached).
Accomplishments that I'm proud of
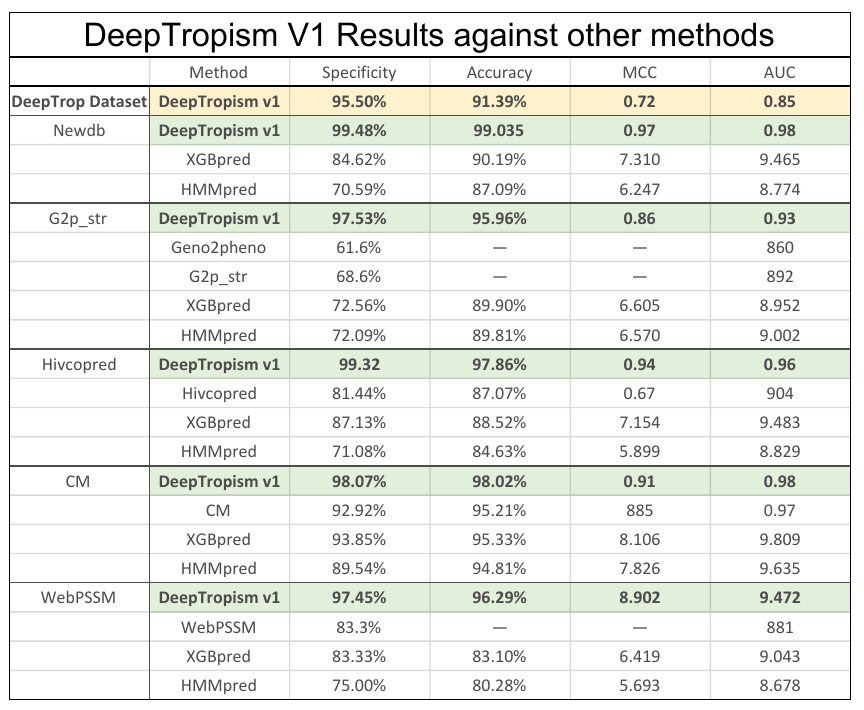
We have created a new Dataset of curated and unique sequences that can be used to train other models of viral tropism. The performance of our DeepTropism Neural Network has surpassed all the published tools in the field (table 1). By using simple feature extraction and architecture we have shown the potential of Pytorch on solving problems that were around for many years and couldn't be tackled with traditional algorithms and tools.
What I learned
Pytorch can be an incredible tool for dealing with any kind of data, even genetic data. I have finally managed to put to test my skills with Pytorch and it was incredibly rewarding to see the results.
What's next for HIV-1 DeepTropism
We do think DeepTropism is a valuable tool for the field of HIV research and drug development and we plan to formalize the results on a scientific article. We also want to create a web tool and API for people to make predictions on other viral sequences. And finally, we encourage other researchers of the field to use Pytorch and the Deep Tropism model to develop other DNN to improve it even more.

Log in or sign up for Devpost to join the conversation.