-
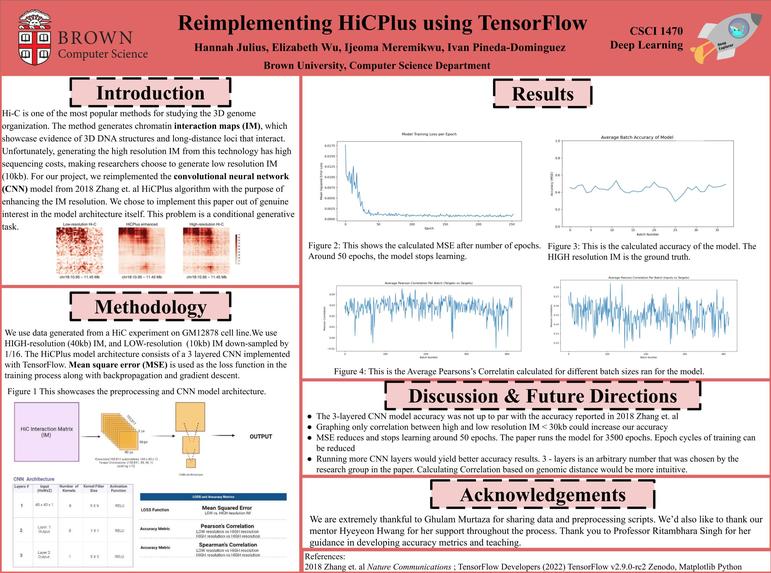
Our poster!
Title: HiC > Fanta
Github: https://github.com/ivan-pd/brown-deep-learning-hic Paper: https://www.nature.com/articles/s41467-018-03113-2.pdf
FINAL REFLECTION:
Link: https://docs.google.com/document/d/1S0V0QMBEuRJ-iSvSChNdYC5tidL5gec9_KwA3S8RPA4/edit?usp=sharing
Names/CS Login:
- Ijeoma Meremikwu (imeremik)
- Hannah Julius (hjulius)
- Elizabeth Wu (ewu32)
- Ivan Pineda-Dominguez (ipinedad)
Introduction:
Hi-C Maps are among one of the most popular methods for studying the 3D organization of the genome. Unfortunately, this technology is plagued by high sequencing costs forcing researchers to use low sampled sequencing ultimately resulting in low-resolution data. To solve this problem, Zhang and colleagues explore the use of convolutional neural networks to enhance the resolution of Hi-C data in their paper: Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus. In this paper, Zhang and colleagues were able to devise a CNN model (HiCPlus) that outperformed other currently used data enhancement methods including 2-D Gaussian smoothing. For our project, we intend to implement the HiCPlus model outlined in the paper and replicate the results demonstrated in the paper using a different dataset. We chose to implement this paper due to the intersection of its content with the interests of members in the group. In addition, the simplicity of the HiCPlus model architecture, which consists of only 3 convolution layers, seemed like a worthwhile and feasible deep learning architecture to implement using the knowledge we have acquired in CS1470. This problem is a supervised learning regression problem, specifically because we are predicting hi-C contact values from noisy inputs.
Related Work:
HiCNN: a very deep convolutional neural network to better enhance the resolution of Hi-C data: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6821373/
In the paper HiCNN: a very deep convolutional neural network to better enhance the resolution of Hi-C data, authors Liu and Wang explore methods for improving the enhancement of Hi-C contact matrices through the use of a very deep CNN. In particular, Liu and Wang proposed a 54 layer CNN architecture for Hi-C enhancement that was inspired by the HiCPlus model we are implementing. The biggest difference between HiCPlus and HiCNN is the number of layers utilized by the CNN architecture. HiCPlus only uses 3 convolutional layers whereas HiCNN uses 54 layers. The first two layers of HiCNN are used for “pattern extraction and representation” and serve the same purpose as the first layer of the HiCPlus architecture. The remaining 52 layers of HiCNN are used for “global and local residual learning” which has been shown to improve the resolution of images enhanced by super-resolution convolutional networks. To evaluate the success of their model, Liu and Wang calculated Pearson correlation coefficients as well as the Mean Squared Error of their low-resolution enhanced Hi-C matrix and that of a high-resolution matrix. The HiCNN architecture was shown to outperform HiCPlus while also reducing training time despite its much larger architecture.
Publicly Available Implementations of HiCPlus: Pytorch Implementation: https://github.com/wangjuan001/hicplus/tree/master/hicplus Deprecated Implementation: https://github.com/zhangyan32/HiCPlus
Data:
We will be using Hi-C Data from GM12878 cells. In particular, we will be using a high-resolution (10kb) Hi-C matrix of GM12878 as well as a 10kb matrix of the same cell with sequencing reads down-sampled by 1/16. If time permits, as part of our stretch goal, we intend to use Hi-C data of K562 and IMR90 cells to replicate results from the paper illustrating that the HiCPlus model can enhance Hi-C data of different cell types when using multiple cell datasets for training.
We found Hi-C data collection outside of what is provided in the paper to be quite difficult. For this reason we are extremely thankful to Ghulam Murtaza for sharing the data and preprocessing scripts he has collected and developed as part of his own research with Hi-C data.
We expect preprocessing and model training to take a long time due to the nature of the data we are using. As mentioned in the HiCNN paper, training the HiCPlus model takes approximately 28 hours using a NVIDIA V100 GPU. With this in mind we intend to use Brown’s high performance computing cluster OSCAR which one of our team members has priority access to use.
Methodology:
The HiCPlus model architecture consists of a 3 layered ConvNet. Conv Layer 1: Pattern Extraction and Representation Input: An NxN low-resolution sample 16 filters each with size 5x5 ReLU non-linear activation function Conv Layer 2: Non-linear mapping between the patterns on high-and low-resolution maps Input: Output from Conv Layer 1 16 filters each with size 1x1 ReLU non-linear activation function Conv Layer 2: Combining patterns to predict high-resolution maps Input: Output from Conv Layer 2 1 filter Note: Mean square error (MSE) is used as the loss function in the training process along with backpropagation and gradient descent.
We will be training our model using Brown’s high performance computing cluster Oscar. We believe that the hardest part of implementing the model will be with regards to image pre-processing our data. In the HiCPlus paper the authors “Divide a Hi-C matrix into multiple square-like sub-regions with fixed size, and each sub-region is treated as one sample”. To replicate this result we expect to spend a significant amount of time figuring out how to divide our Hi-C matrix to provide good inputs to our CNN model.
Metrics:
In the HiCPlus paper, Zhang and colleagues implemented multiple different tests to show the utility of a CNN based Hi-C data enhancer. The authors demonstrated that chromatin interactions are predictable from neighboring cells using a CNN, that it is possible to enhance a chromatin interaction matrix with low-sequence depths, that Hi-C interaction matrices can be enhanced using HiCPlus across different cell types, and that its possible to identify chromatin interaction from HiCPlus-enhanced matrices. For the purpose of this project we will focus on predicting chromatin interaction from neighboring cells and enhancing chromatin interaction matrices with low-sequence depths as our base and target goals respectively. As a stretch goal we hope to use our HiCPlus implementation for Hi-C data enhancement across different cells. However, we predict that our stretch goal will require more time with regards to data collection, preprocessing, and training that might not be feasible to complete within our given timeframe.
In order to measure the accuracy of our HiCPlus implementation we plan to implement the same measures of accuracy as outlined in the paper. Primarily, we will be using Pearson and Spearman correlation coefficients between the predicted HiCPlus values and the real values of our high-resolution HiC-Data. As an alternative, we can also use Mean Square Error as a measure of accuracy as shown in the HiCNN paper by Liu and Wang .
Ethics:
Who are the major “stakeholders” in this problem, and what are the consequences of mistakes made by your algorithm? Major stakeholders include genomic researchers who utilize tools like Hi-C Maps for genomic research and analysis. The utility of using a CNN to enhance Hi-C data rests on the ability of the model to accurately predict cell values in Hi-C Maps with low-resolution or noisy data caused by down-sampled sequencing. The consequence of the algorithm making a mistake in Hi-C cell prediction leads to inaccurate results and interpretation of data by genomic researchers. Due to the sensitivity and importance of genomic research in curing new diseases, learning more about the genome, and potentially creating new medications from genomic research this could result in grave consequences. In addition, researchers are trying to get high quality data through new developed methods. HiC experiments require high deep sequencing in order to obtain high resolution. This is a costly venture both in time and economics. Therefore, if we are able to obtain high resolution data based upon low-resolution results generated from chromatin capture experiments.
Why is Deep Learning a good approach to this problem? On a high level, the problem is an image resolution problem, as we want to infer high-res Hi-C matrices from low-res Hi-C samples, essentially taking a low-res image and increasing the resolution. Neural networks are great for this, because they can learn how to remove corruption such as noise from input images, ultimately producing a restored, higher-resolution image. Convolutional autoencoders in particular are good at upscaling image resolution, as you can use matrix multiplication with the transpose of a larger convolution matrix and the flattened input image to increase the size of the output vector, ultimately creating a larger output image based on the feature scoring of the convolution matrix and the original image.
Division of labor:
- Data Collection and Preprocessing Thank you again to Ghulam Murtaza for sharing the data and preprocessing scripts Ijeoma Meremikwu
- Developing Model Architecture Model.py: Elizabeth Pred chromosome: Ijeoma, Ivan Pred genome: Hannah, Ijeoma Utils: Everyone [tentative]
- Training and Testing Model Training: Elizabeth, Hannah Testing: Ijeoma, Ivan
- Data Analysis/Visualization of Results (Creating Figures) Ijeoma, Ivan Accuracy Pearson’s R correlation Spearman R correlation
- Poster Elizabeth Wu Hannah Julius
Built With
- python
- tensorflow

Log in or sign up for Devpost to join the conversation.