
Inspiration
We both know people who’ve cycled through three, four, five psychiatric medications before finding one that works. The pattern is always the same: try a drug, wait 6–8 weeks, experience side effects, switch, repeat. It can take over a year to reach remission — and for some people, it never happens.
Pharmacogenomics has started to address this by testing patients’ liver enzyme variants (CYP2D6, CYP2C19), but it only explains part of why the same drug works for one patient and fails for another. We kept asking: what explains the rest?
The answer, increasingly, is the gut. Every oral medication passes through a gastrointestinal tract colonized by trillions of bacteria — and those bacteria have their own drug-metabolizing enzymes. Zimmermann et al. (2019, Nature) screened 76 gut bacterial species against 271 drugs and found that two-thirds were significantly metabolized by gut bacteria through oxidoreductases, hydrolases, and nitroreductases. This is well-established science. But no clinical tool connects a specific patient’s microbiome profile to their psychiatric drug options.
That’s what Gut Instinct does.
What It Does
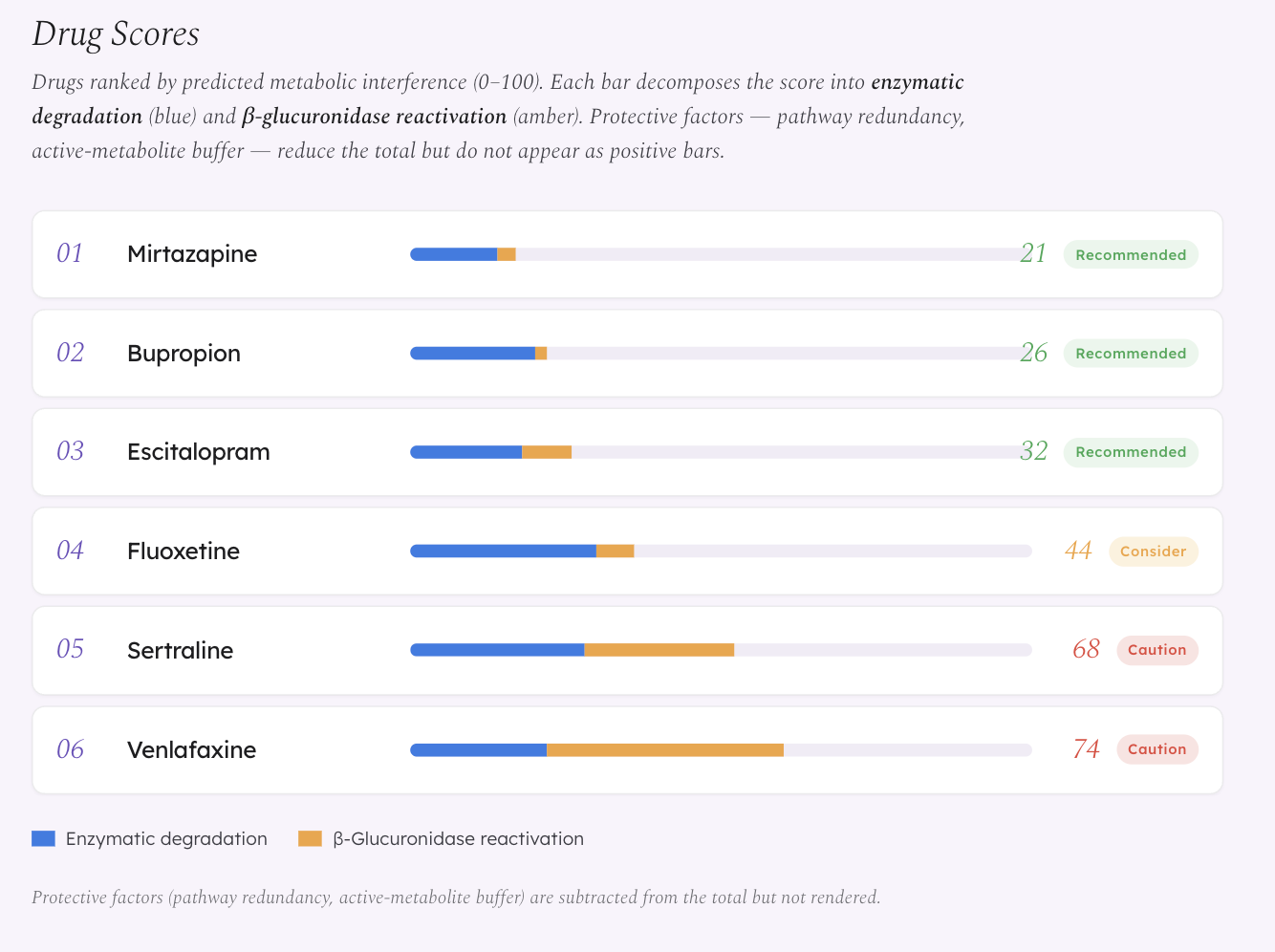
Gut Instinct is a computational framework that maps a patient’s gut microbiome composition against psychiatric drug pharmacokinetics to predict which antidepressants face the least metabolic interference from the patient’s specific bacterial community.
We model two primary mechanisms:
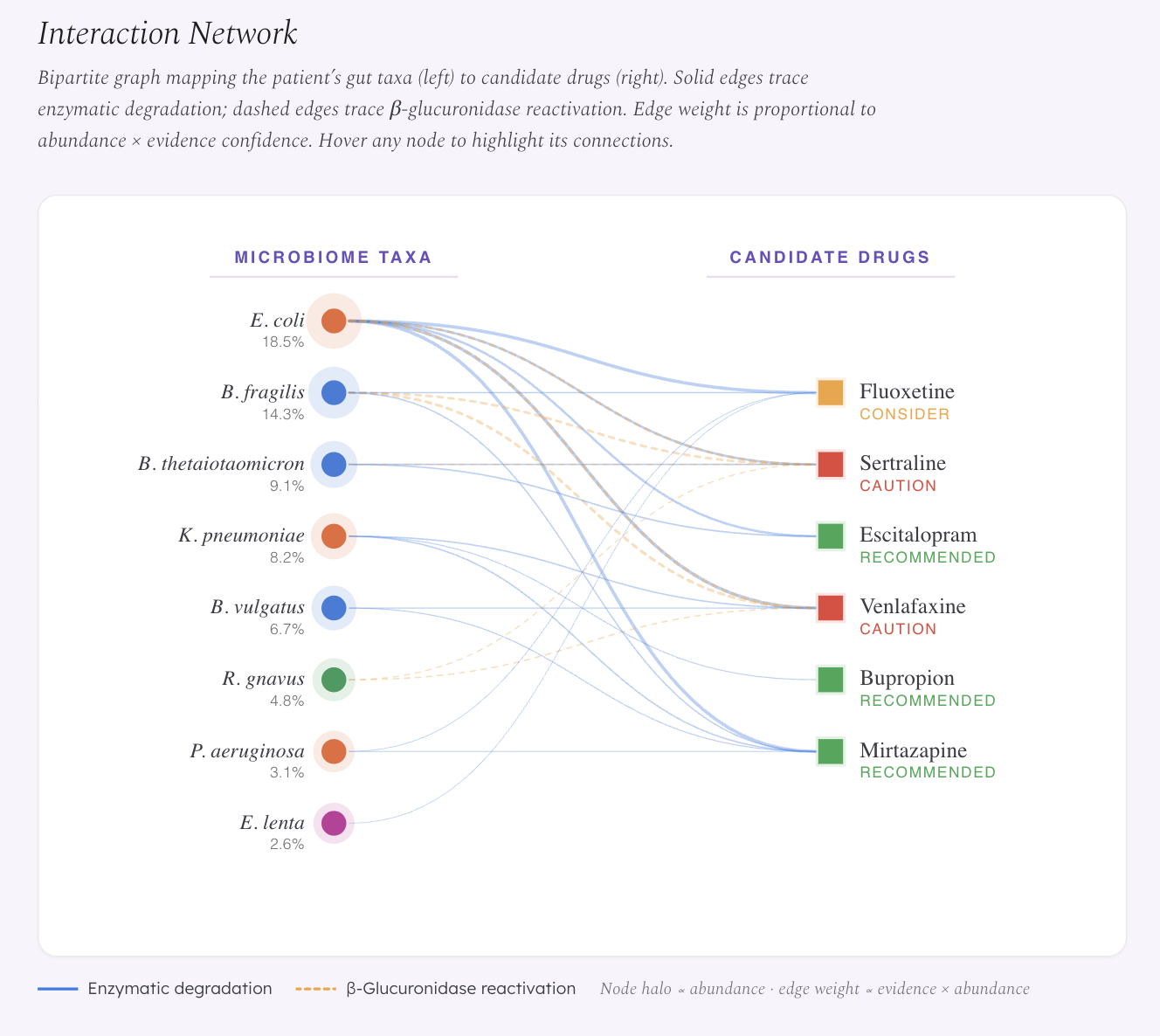
Enzymatic degradation — gut bacteria carry oxidoreductases and hydrolases that can break down drugs in the gut lumen before they reach systemic circulation.
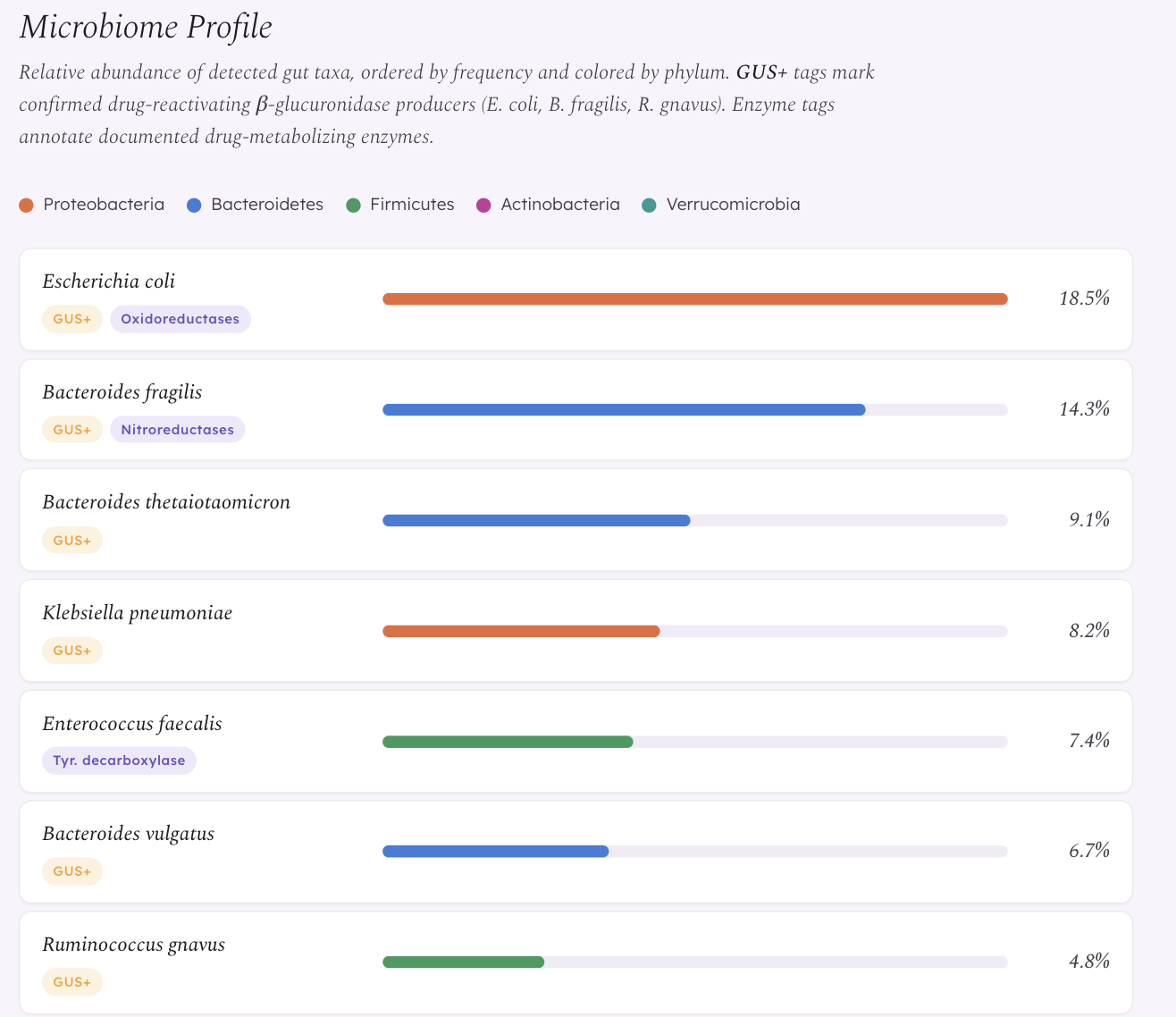
β-Glucuronidase (GUS) reactivation — drugs that the liver has already deactivated via glucuronidation are excreted into the gut, where bacterial GUS enzymes cleave the glucuronide conjugate and release active drug back into circulation. This creates unpredictable plasma levels. This mechanism is already experimentally confirmed in oncology — bacterial GUS reactivation of irinotecan’s metabolite SN-38 causes dose-limiting diarrhea (Wallace et al. 2010). We extend this framework to antidepressants that undergo glucuronidation, particularly Venlafaxine, whose active metabolite ODV is primarily eliminated via this pathway.
The platform runs four AI agents in sequence:
- Agent 1 (Pharmacokinetic Mapper) extracts CYP enzyme pathways, protein binding, and glucuronidation flags for each candidate drug
- Agent 2 (Enzyme Matcher) identifies which bacteria in the patient’s gut carry documented drug-metabolizing enzymes
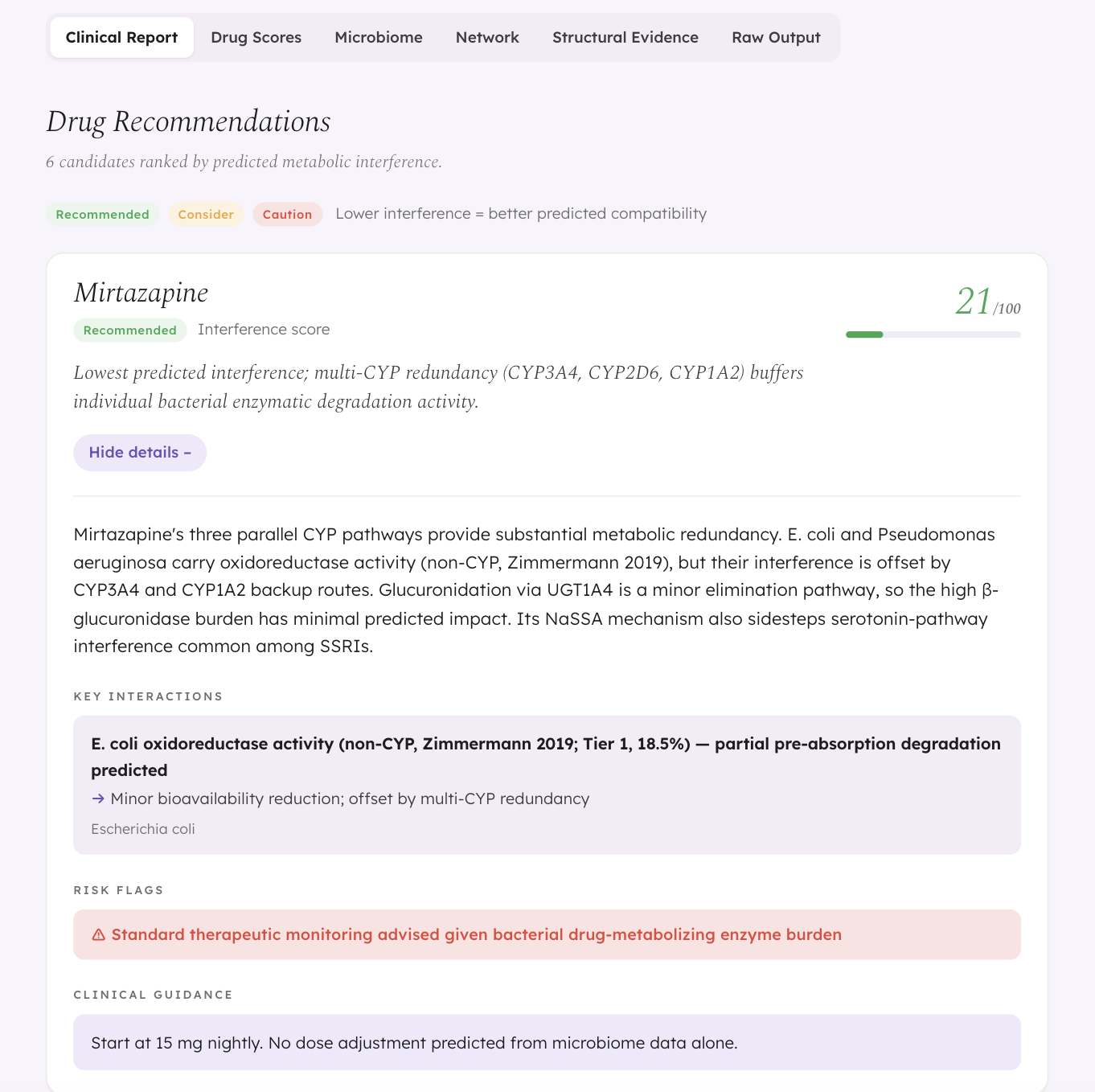
- Agent 3 (Graph Architect) builds a knowledge graph and computes a metabolic interference score (0–1) for each drug
- Agent 4 (Clinical Interpreter) translates scores into tiered, clinician-readable recommendations
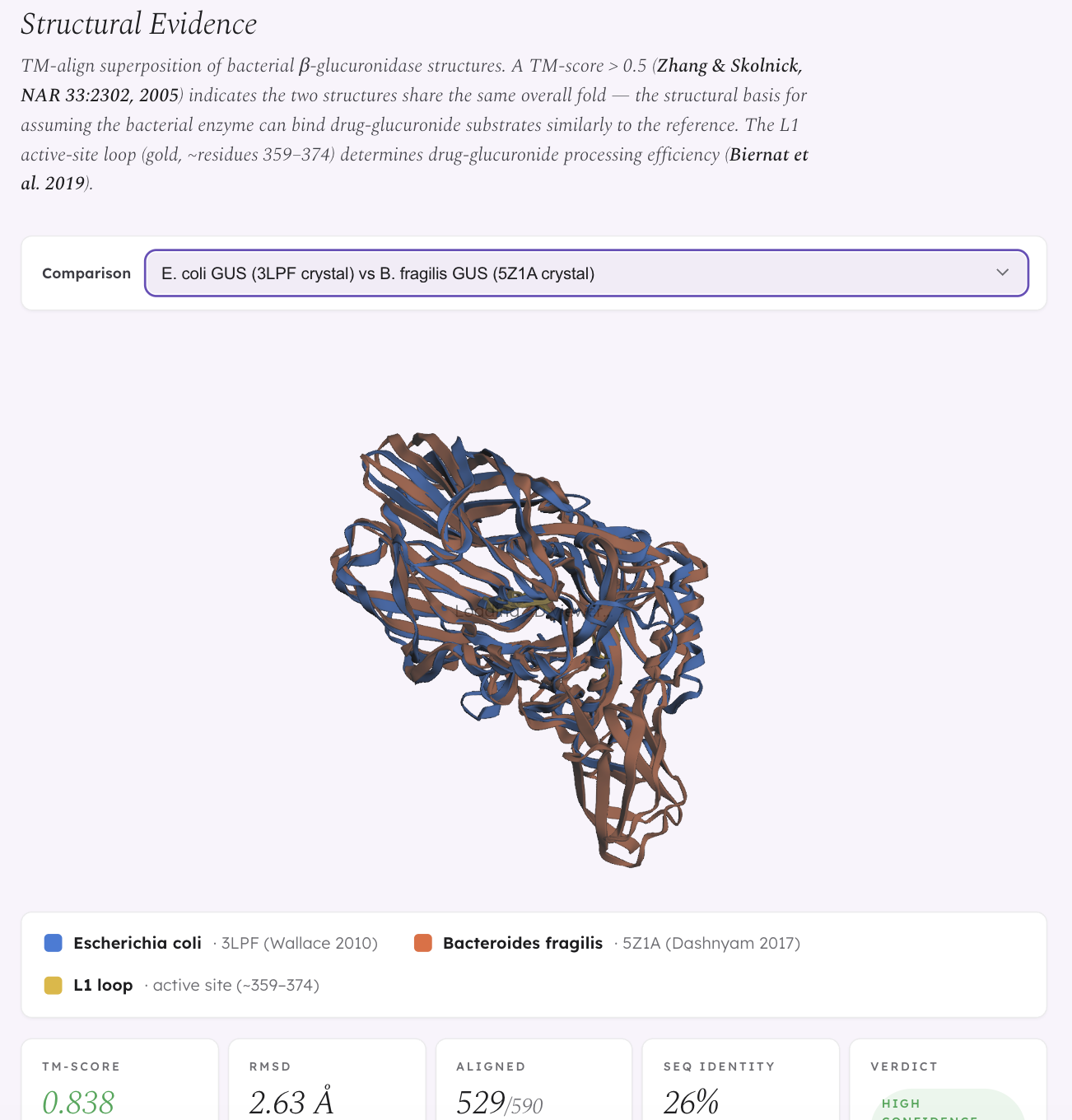
We also built a structural validation layer using TM-align superposition of bacterial β-glucuronidase crystal structures (from PDB) and AlphaFold predictions. E. coli GUS and R. gnavus GUS are structurally near-identical at the fold level (TM-score 0.949) — yet Biernat et al. (2019, Scientific Reports) showed that R. gnavus GUS processes drug substrates less efficiently. This means global structural similarity alone doesn’t predict functional equivalence; subtle active site differences drive differential drug processing, which is exactly why structural validation matters beyond a simple “GUS present = risk” model.
The key demo finding: same diagnosis, different microbiome, different best drug. A patient with MDD and post-antibiotic dysbiosis gets Mirtazapine as the top recommendation (score 0.21). A different MDD patient with IBS-associated Firmicutes dysbiosis sees Mirtazapine drop to 0.52 — because their enriched Clostridium and Bacteroides populations create broader enzymatic pressure across multiple metabolic pathways. Same drug, same diagnosis, different microbiome, different recommendation.
How We Built It
- Backend: FastAPI (Python) orchestrating four sequential Claude API calls, each with a curated pharmacomicrobiomics knowledge base as the system prompt. Deployed on Railway.
- Frontend: React (CDN) with a custom dark-mode clinical UI. SVG-based bipartite interaction network. Deployed on GitHub Pages.
- Structural validation: Downloaded GUS crystal structures from PDB (3LPF, 5Z1A) and AlphaFold predictions (AF-Q6W7J7-F1 for R. gnavus). Ran TM-align to compute structural similarity scores. Built a 3Dmol.js interactive viewer to visualize superposed structures with highlighted L1 active-site loop regions.
- Biological fact-checking: We manually verified every enzyme claim against UniProt, PDB, and primary literature. This led to a major correction — we discovered that E. coli and B. fragilis do NOT carry native CYP450 genes (zero UniProt results), and that E. faecalis produces beta-galactosidase, not beta-glucuronidase. We corrected the entire knowledge base to reflect the actual enzyme families documented in Zimmermann et al. 2019.
Challenges We Ran Into
The CYP ortholog problem. Our initial knowledge base described bacterial enzymes as “CYP2D6-like” and “CYP3A4-like” — language borrowed from early pharmacomicrobiomics literature. When we fact-checked against UniProt, we found zero native CYP450 genes in E. coli or B. fragilis. The drug metabolism is real, but the enzymes responsible are oxidoreductases and hydrolases, not CYP orthologs. We had to rewrite the entire knowledge base and every piece of frontend text that referenced CYP orthologs. This was painful but made the science defensible.
Structural validation integration. Getting TM-align output into a browser-renderable 3Dmol.js viewer required converting superposed PDB coordinates, mapping L1 active-site loop residue ranges for each organism, and handling the fact that crystal structures and AlphaFold predictions have different chain naming conventions.
Balancing rigor with usability. A psychiatrist doesn’t need to see TM-scores and evidence tiers — but a researcher evaluating the framework does. We built the UI with progressive disclosure: the Clinical Report tab gives a clean recommendation, while Drug Scores, Interaction Network, Structural Evidence, and Raw Output tabs provide full transparency for anyone who wants to interrogate the biology.
Accomplishments We’re Proud Of
- Every biological claim in the platform is now traceable to a specific UniProt ID, PDB structure, or peer-reviewed paper
- The fact-check itself — finding and fixing the CYP ortholog error took intellectual honesty, not just engineering skill
- Four demo cases producing four different drug rankings, demonstrating that the system captures real biological variation rather than always recommending the same drug
- A working 3Dmol.js structural viewer showing superposed GUS enzymes with quantitative TM-scores
What We Learned
The most important lesson was that biological plausibility is not biological accuracy. “E. coli carries CYP2D6-like orthologs” sounds reasonable and appears in secondary literature — but when we checked the primary databases (UniProt, PDB), the claim fell apart. The drug metabolism is real (Zimmermann 2019 confirmed it experimentally), but the mechanism is through entirely different enzyme families. Building a clinical decision support tool on unchecked claims would be irresponsible. The fact-check made the tool slower to build but fundamentally more trustworthy.
We also learned that structural biology reveals surprises that sequence-level or taxonomy-level analysis misses. E. coli and R. gnavus both carry β-glucuronidase with nearly identical folds (TM-score 0.949), yet they process drug substrates at different rates (Biernat et al. 2019). Structural similarity at the global level doesn’t guarantee functional equivalence — the differences that matter are in the active site geometry, not the overall fold.
What’s Next
- Experimental validation: The core hypothesis — that bacterial GUS enzymes cleave antidepressant-glucuronide conjugates — needs in vitro confirmation. A future assay using recombinant E. coli GUS (P05804) with Venlafaxine/ODV-glucuronide as substrate would directly test this.
- Expanding the drug panel: The current knowledge base covers 6 antidepressants. Extending to anxiolytics, antipsychotics, and mood stabilizers would broaden clinical utility.
- Integrating pharmacogenomics: Combining microbiome-based predictions with CYP2D6/CYP2C19 genotype data would create a more complete picture of individual drug metabolism variability.
- Modeling bidirectional effects: SSRIs like fluoxetine have antimicrobial activity against gut bacteria. The current model treats the microbiome as static — a future version should account for the drug reshaping the very microbiome that metabolizes it.
References
- Zimmermann, M. et al. (2019). Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature, 570, 462–467.
- Wallace, B. D. et al. (2010). Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science, 330, 831–835.
- Biernat, K. A. et al. (2019). Structure, function, and inhibition of drug reactivating human gut microbial β-glucuronidases. Scientific Reports, 9, 825.
- Haiser, H. J. et al. (2013). Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science, 341, 295–298.
- Maini Rekdal, V. et al. (2019). Discovery and inhibition of an interspecies gut bacterial pathway for levodopa metabolism. Science, 364, eaau6323.
Built With
- 3dmol.js
- alphafold
- claude-api
- fastapi
- github
- html
- javascript
- pdb
- python
- railway
- react
- tm-align

Log in or sign up for Devpost to join the conversation.