-
-
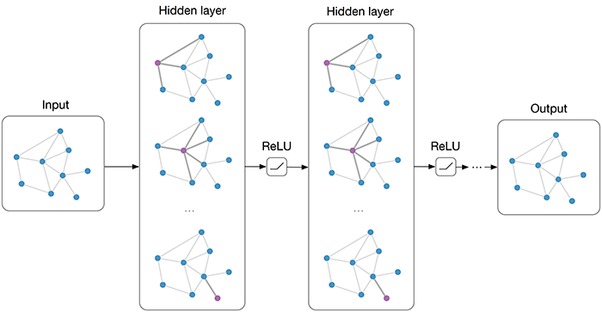
Graph Neural Network Message-Passing Phase (before Readout phase)
-
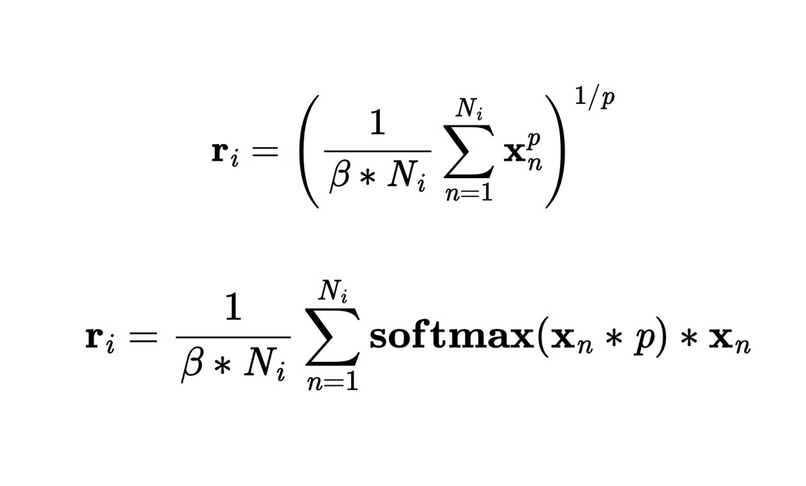
The 2 families of Generalized Mean-Max-Sum functions used in the novel Readout layer
-
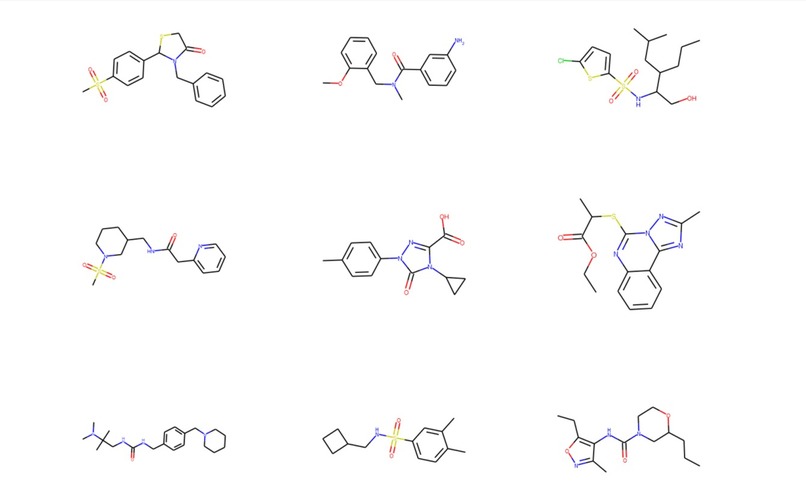
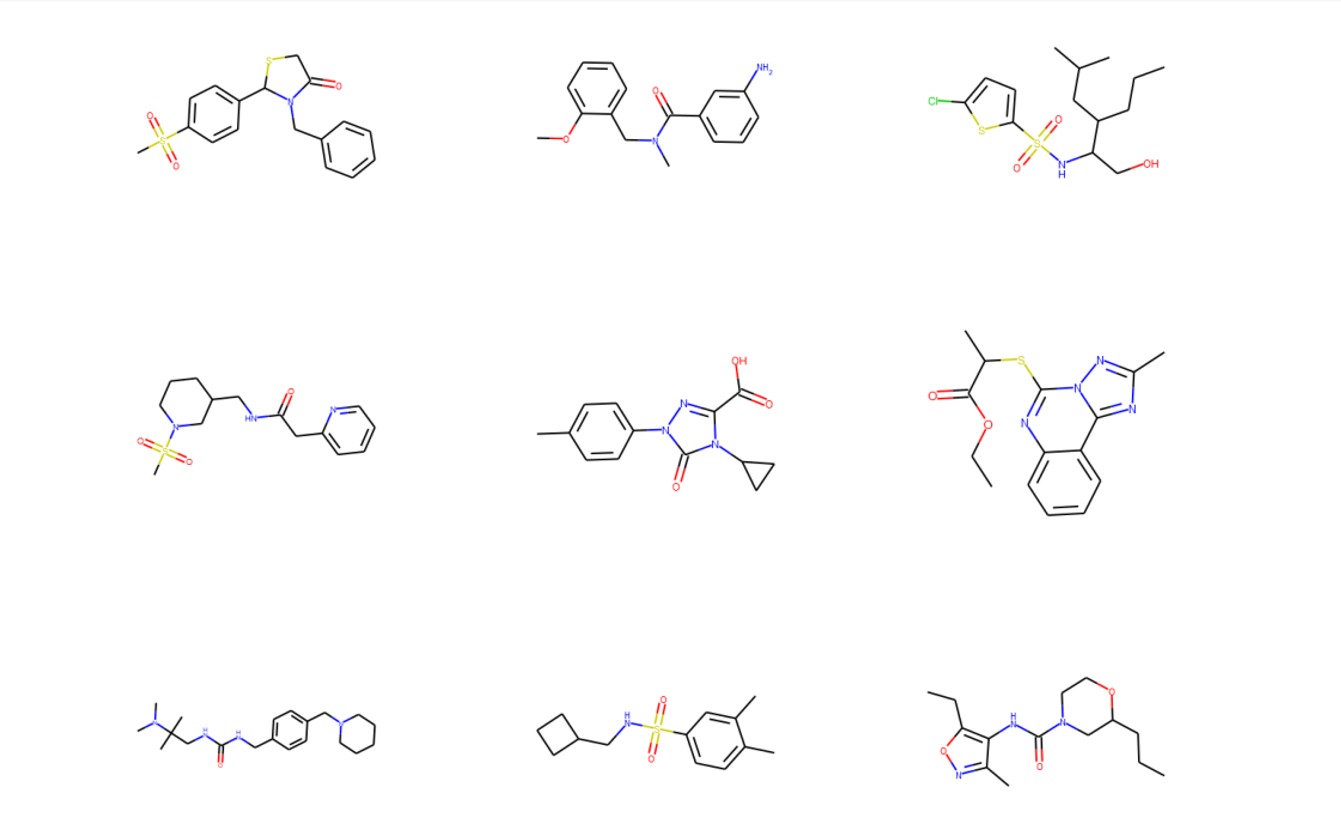
Sample of Molecules produced by MolDSM (our generative model)
-
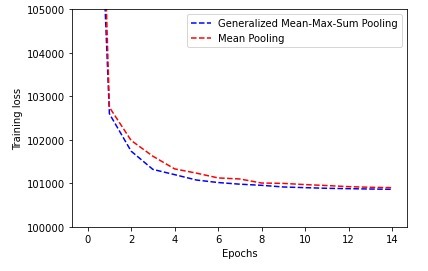
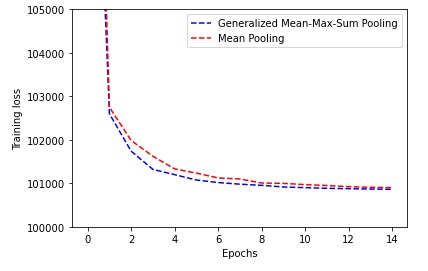
Training Loss (traditional mean pooling vs generalized mean-meax-sum) evolution (lowerr is better)
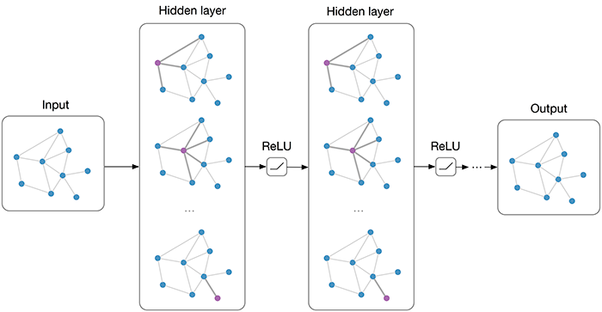
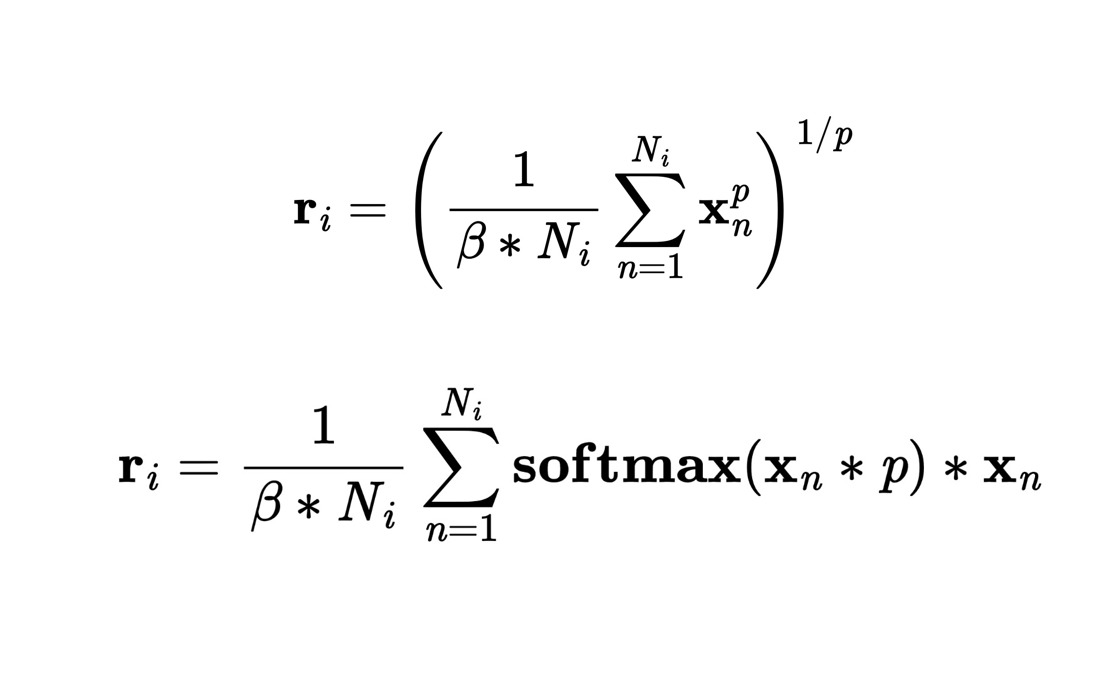
Inspiration
A new type of generative models (AIs that can generate different things) was proposed last year by a Stanford PhD. Two months ago, the same author released an improved implementation solving some problems of the initial model. In parallel, Graph Neural Networks have seen a substantial improvement in the recent years (from 2015 until now). Given my background in Medicine, Physics and AI, I decided to implement the newest (and thus the best) GCN architecture I could find together with the new generative model framework.
What it does
I applied a Graph Convolutional Network trained under the scheme of Denoising Score Matching (DSM) with Langevin Sampling (LS) to the molecular generation problem with an underlying permutation-invariant representation of molecules. The model consists of a modification of a very recent Graph Convolutional Network (GCN) architecture with a novel readout phase consisting of a Generalized Mean-Max-Sum global pooling function. The preliminary results show that our framework has good molecular generation performance when compared to existing ones and it could also be used in a molecular optimization scheme via reinforcement learning.
How I built it
To build this project, I used the description of the implementations described in the original 2 papers:
- Generative models: Improved Techniques for Training Score-Based Generative Models (Jun 16, 2020)
- Graph ConvNet: DeeperGCN: All You Need to Train Deeper GCNs (Jun 13, 2020)
I implemented the model in the PyTorch and PyTorch Geometric libraries, and I've used the RDKit software package for the cheminformatics tasks. The project has been developed iteratively in different jupyter notebooks.
Challenges I ran into
Implementing very recent papers always has issues. The most important ones have been:
- Lack of documentation and/or community: there's no one to ask since it's at the frontier of Deep Learning / AI research. That means no StackOverflow, no Github issues...
- Lack of examples: I was doing something no one has done ever (not even close) so I had to think about all the algorithms myself. There wasn't any reference implementation in many cases.
- Lack of measures:
- Computational Constraints: I developed this project in my own PC, whereas the Stanford PhD that proposed the generative framework used the High Performance Computing Cluster from his University. This has been an important klimitation since Denoising Score Matching - Langevin Sampling is a very computational-intensive framework.
Accomplishments that I'm proud of
Although it might be easy to understand the project as just a Generative Model that produces molecular graphs, I had to program several algorithms to bring it to life, including:
- Graph encoding of molecules based on the different atoms and bonds (converted to nodes and edges)
- Graph decoding (from nodes and edges to a representation that can be visualized )
- Continuous encoding of molecules (this one was hard): DSM-LS (the model framework) needs a continuous representation to work with, and graphs are not continuous by default.
- Continuous decoding of molecules since we needed to retrieve the generated structures.
- Graph Neural Network implementation: i used avery recent paper from Jun 13, 2020 that I had to implement.
- Generalized Readout Layer: I came up with an improvement over traditional Readout functions, so we had to program it.
What I learned
This weekend has been certainly a challenge and I'm proud of the things I did and I learned to do, the most important of which are:
- Working with the PyTorch and PyTorch-Geometric libraries for Deep Learning since I was used to the Tensorflow and Keras libraries, but not PyTorch.
- Working with Graph-structured data and Graph Neural Networks (I had worked with Convolutional Networks for images, Sequence and Tabular data before, but never Graph-like data and Graph ConvNet architectures)
- Efficiently implement complex algorithms from scratch that work
- Doing complicated stuff under an ambitious deadline works.
What's next for Graph Denoising
- I'm preparing a submission of a quick paper to a PrePrint server (I'm considering Arxiv or ChemRxiv). However, the model has 3 minor things that I still need to check:
- Improving the rate of valid molecules: right now it's around 60%-90% but it's sensitive to the training dataset. The ideal rate should be 90-100%. I have to embedd the validity constraints into the algorithm to ensure that the generated molecules are valid by default.
- Improving efficiency: the generation of new molecules takes 6 seconds / molecule on a single CPU core (the annealed langevin dynamics for the sampling are expensive). Right now, I can reduce the time per molecule by parallelizing the sampling accross different CPU cores, but a more in-depth study of the langevin dynamics process could reduce the time/molecule on a single CPU core by 50% approx.
- Benchmarking the improvements against existing methods (I didn't have time to do that in just 1 day!)


Log in or sign up for Devpost to join the conversation.