-

HUVEC cell-line co-expression data (subset)
-
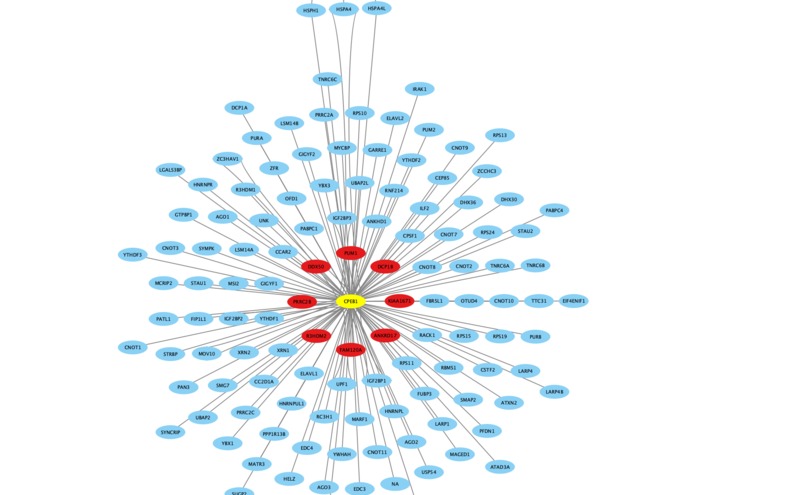
PPI network for one bait in HEK293 cell line
-
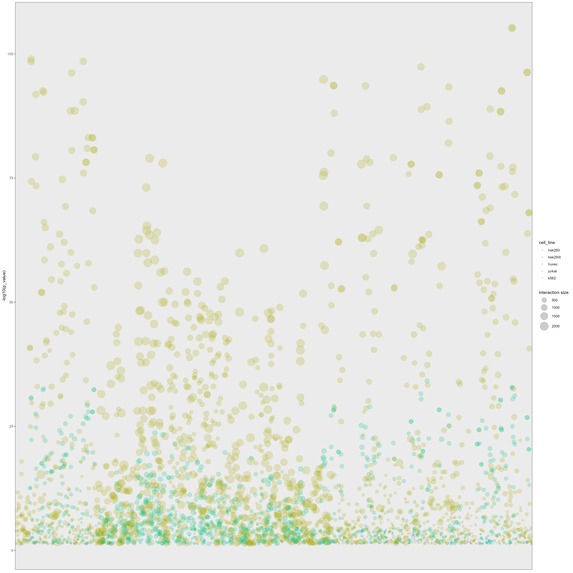
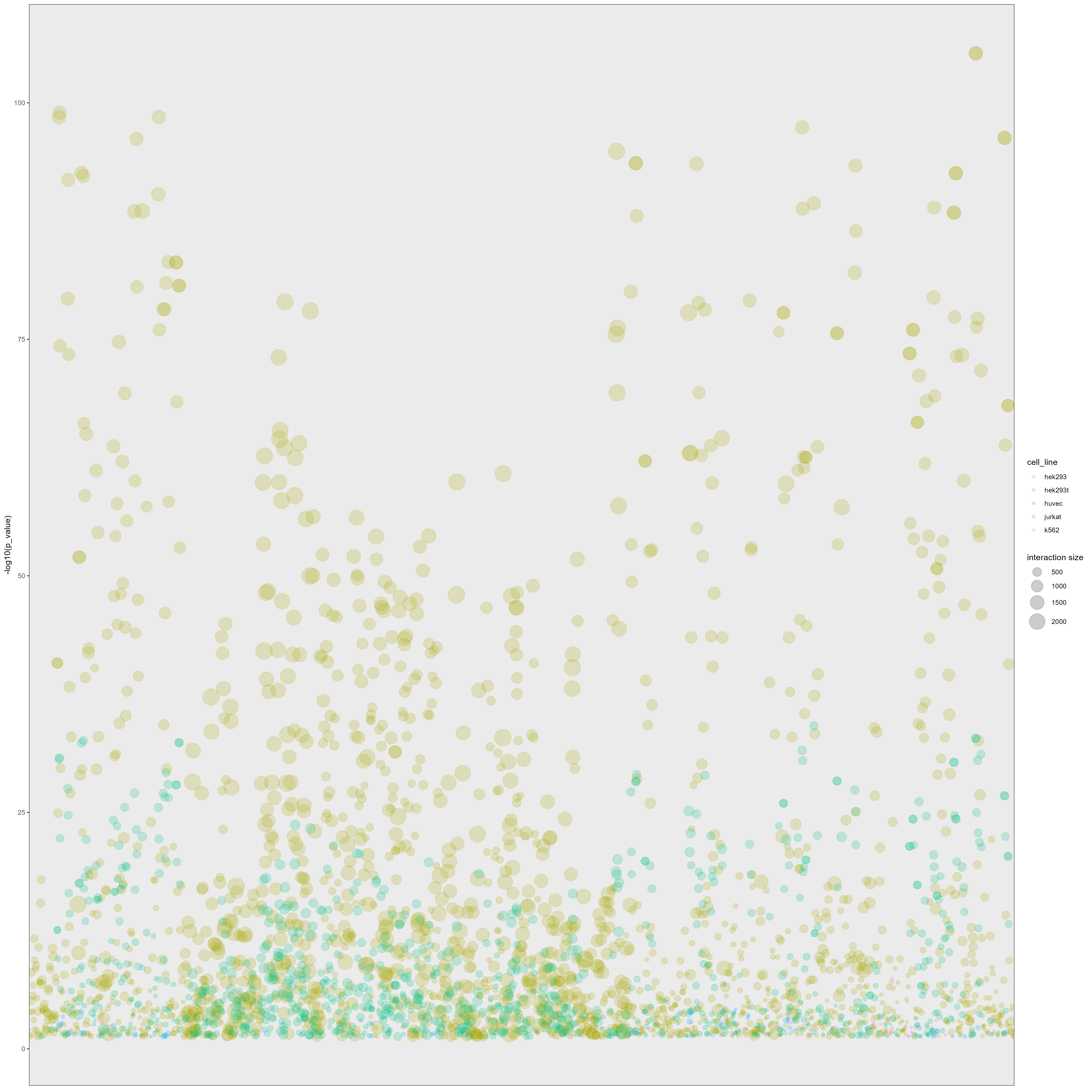
Bubble plot of GO enrichment analysis of predictive proteins
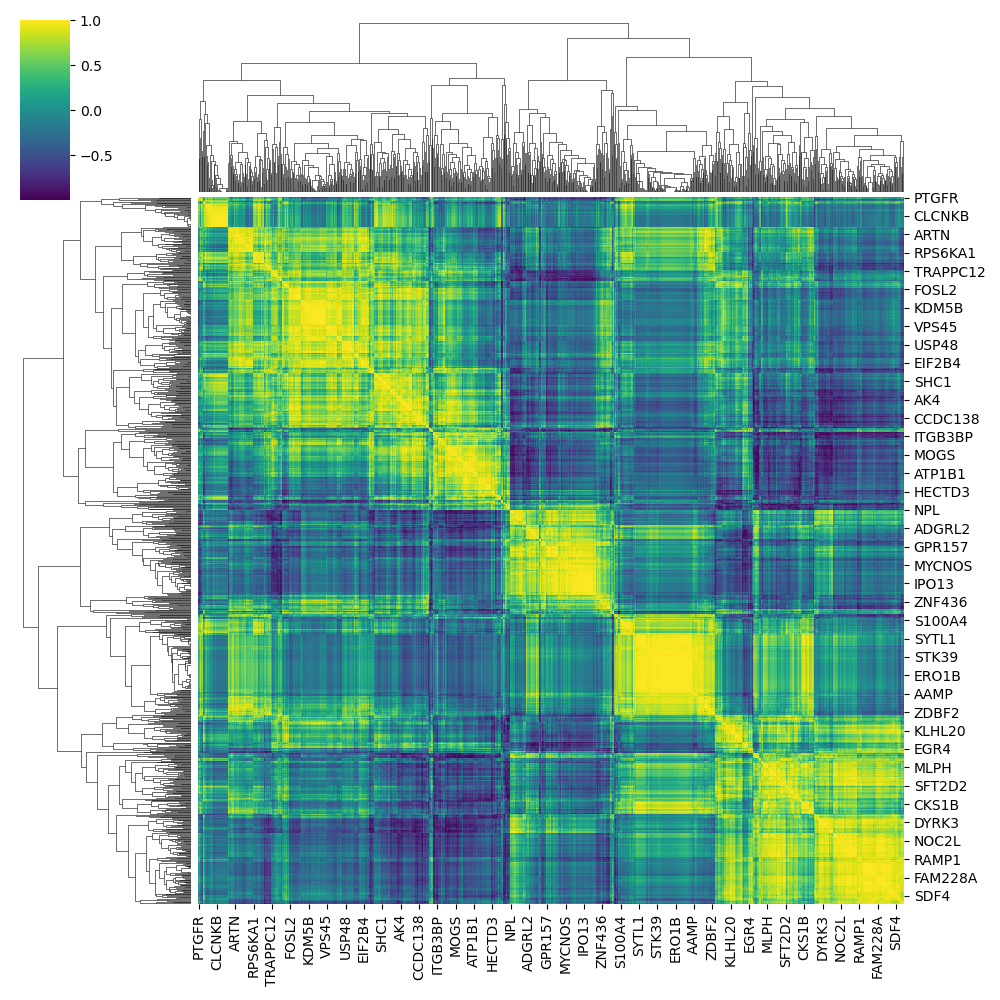
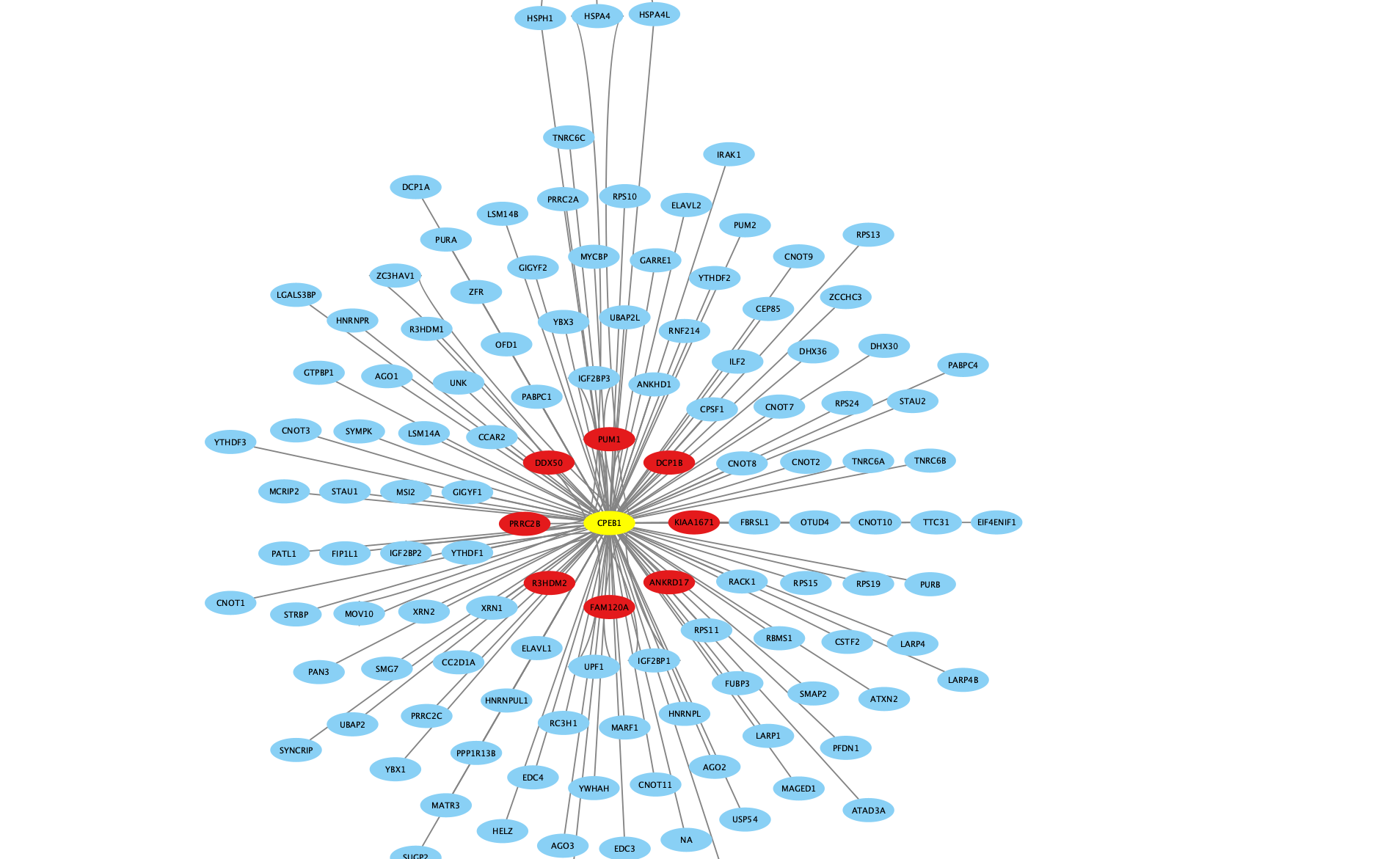
Inspiration
Transcriptomics data is common for many biological contexts, but proteomics data is rare. As such, characterizing protein-protein interactions (PPIs) of disease states is difficult, slow, and expensive. We hope to leverage signals in protein-encoding transcriptomics data as a predictive tool to associate with relevant PPI networks, providing insight into modality with less comprehensive data.
What it does
Our project is built upon two components. First, we generate a co-expression matrix of transcriptomics data on commonly used human cell lines. Then, on the same cell lines we obtain a ranked set of PPIs. Our workflow measures the predictive power (by ROC) of the top-k set of co-expressed protein-encoding genes from the transcriptomics data in determining an analogous set of PPIs.
How we built it
Our project is built on a set of R and Python scripts that transform .csv data files taken from publicly available datasets (GEO, IMEx, PROPER, EMBL-EBI). Inputs and outputs between scripts are defined as mutually intelligible DataFrames between both of our programming languages. Computational work was done across a range of platforms: the hackathon's allocated cluster, our members' own lab allocations, and personal computers.
Challenges we ran into
Spotty availability of the cluster and inability to configure an environment in the same shared setting meant that we were unable to set up a system in which every member was able to concurrently work on the same large data files in the same shared space. Therefore, we had to improvise and create a workflow in which each member worked on different pieces and types of data that were manageable for their environment, and coordinated a central version control system via Github to coordinate all our contributions. We also had issues with non-standard metadata when pulling data online, while other datasets required for either transcriptomics/proteomics data were frequently inaccessible. Due to the limited data we were working with, the predictive signal we were getting was too weak to perform a genuine machine learning experiment as originally planned, and we reduced the scope of our work accordingly.
Accomplishments that we're proud of
We're proud of getting a (weak) predictive signal from RNA co-expression to PPI data despite our limited datasets. We were able to perform novel analysis on an underexplored type of multi-modal data, and our results demonstrate that there is the possibility that with more time and resources available, a ML project on such a topic would be feasible. While we were unable to concurrently work in the same file system, we were able to establish a compartmentalized workflow where we shared intermediate results with each other, and this workflow allowed us to meet most of our project goals in an organized and coordinated manner.
What we learned
We learned that gene co-expression relationships translate pretty weakly to PPI predictions. The presence of some signal in our findings suggest that there is variability across proteins and cell lines in how well the PPIs map back to gene-encoding RNAs. We obtained a conservative baseline performance of this kind of correlative model, given minimal data to work with, and suspect that a larger, more diverse, and more robust multi-modal dataset will enable the development of supervised learning models to create accurate predictions on stronger signals.
What's next for Co-expression Bridge
We wish to publish the methods used in our project as a set of importable and installable packages that others can use to analyze their own data. Obtaining more datasets would enable us to up-scope our project accordingly and approach our originally envisioned project goal of training a model to perform RNA-seq to PPI predictions.

Log in or sign up for Devpost to join the conversation.