-
-
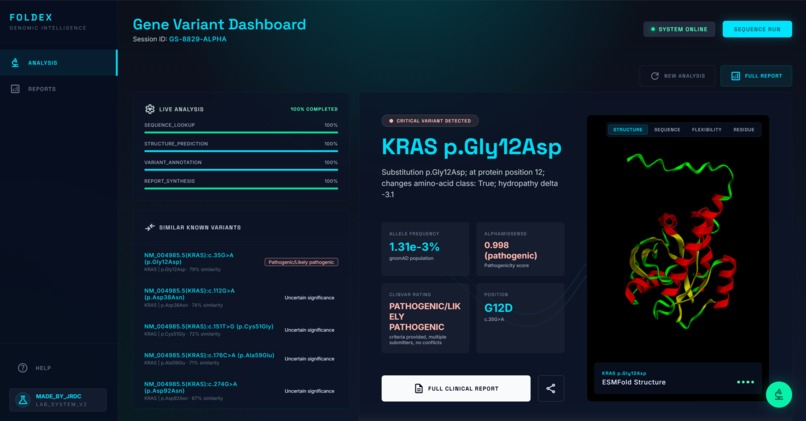
Analysis Page
-
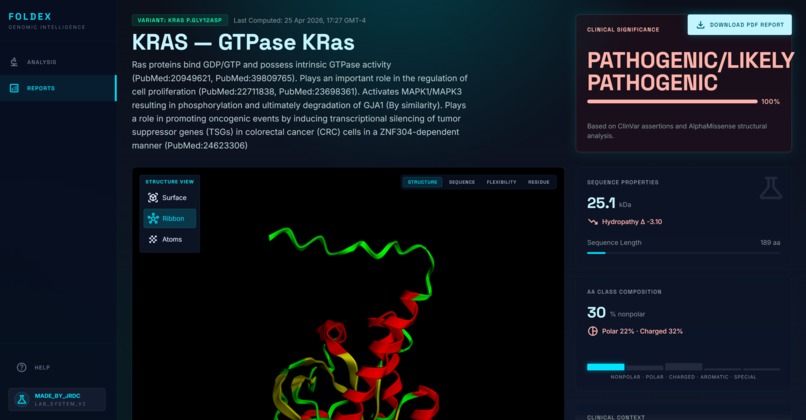
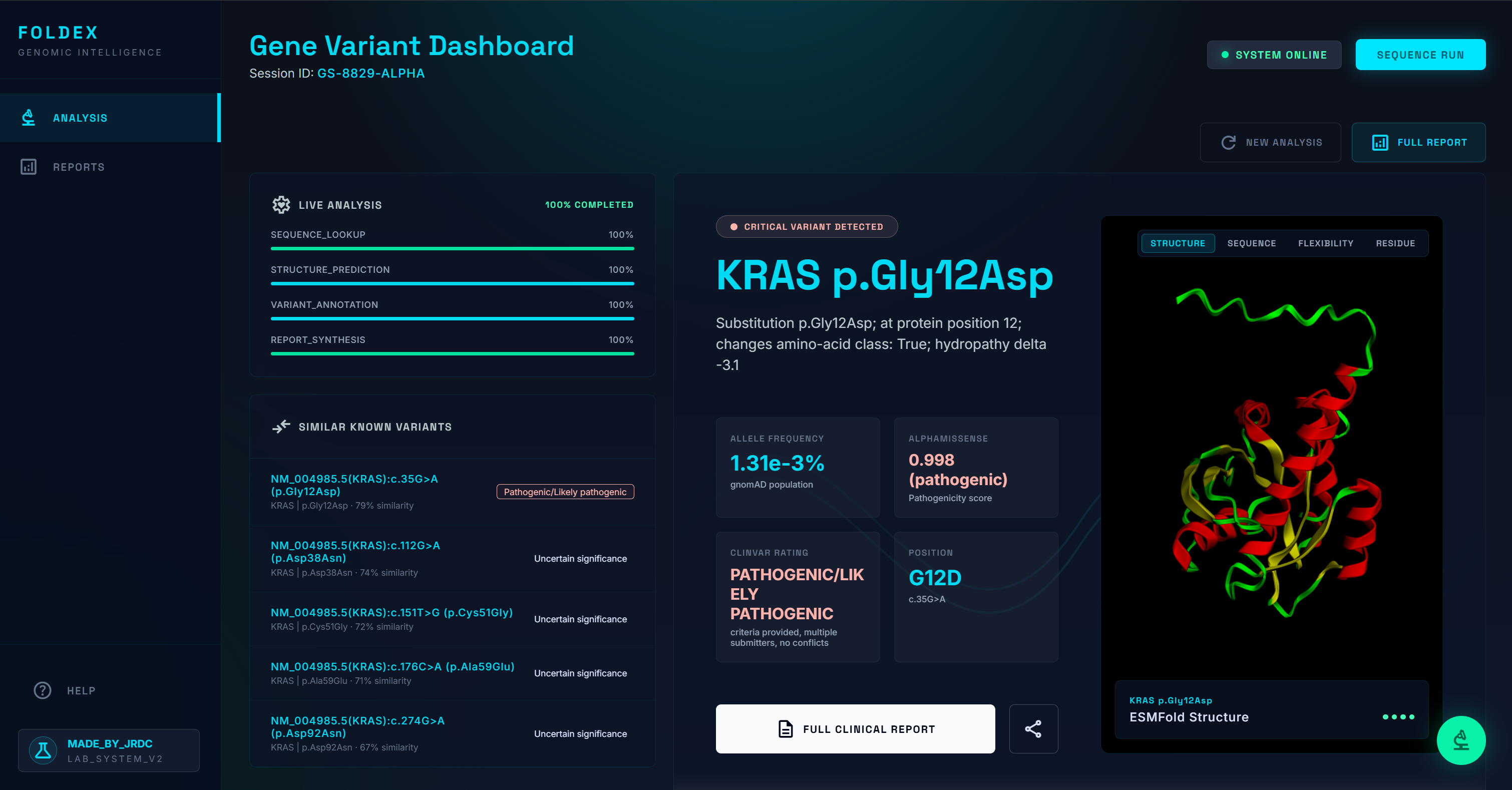
Variant Description
-
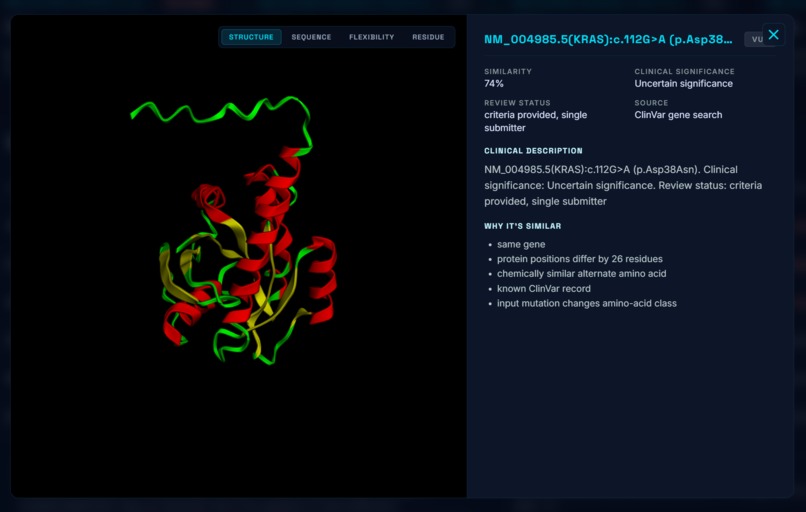
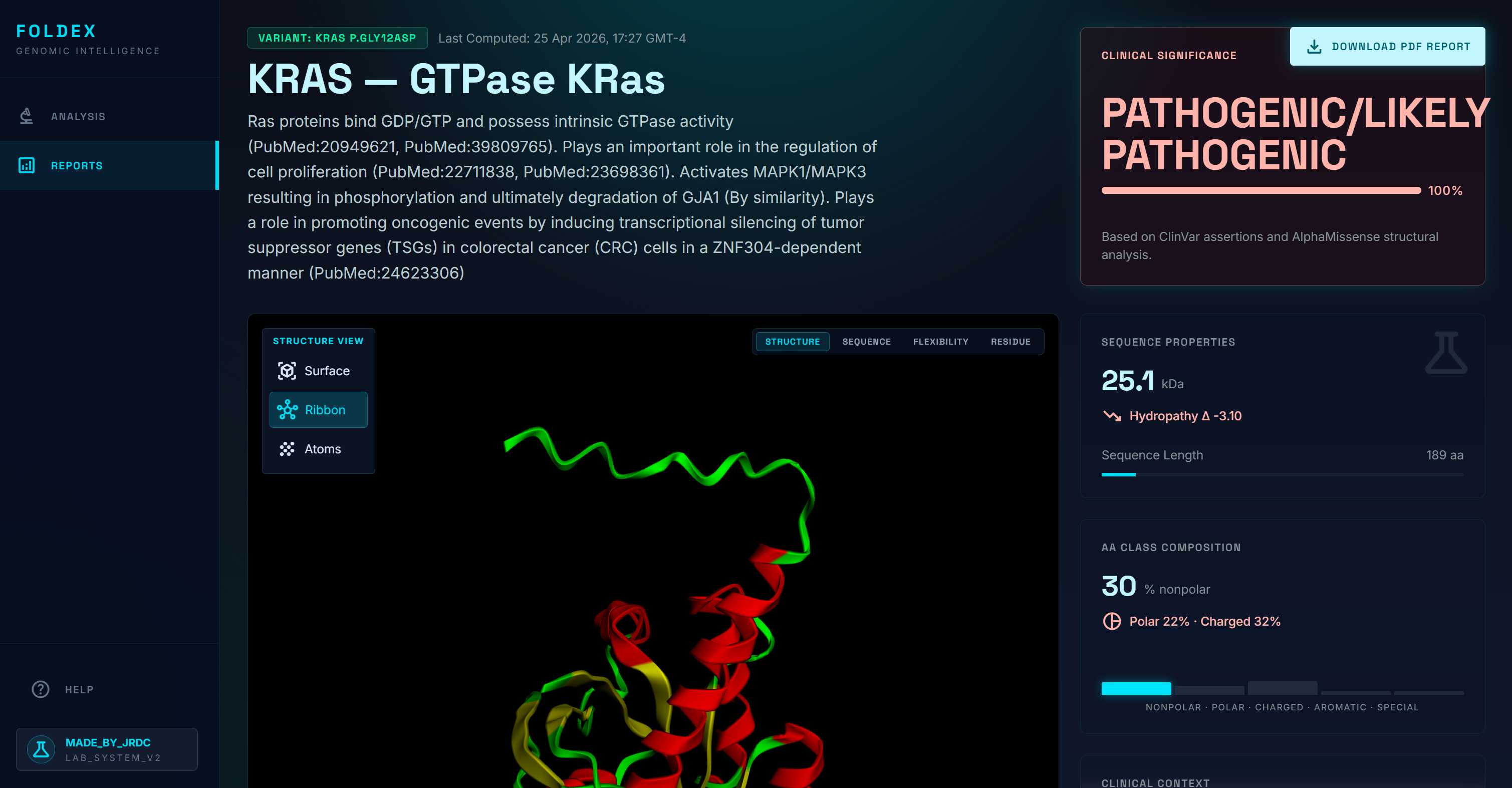
Protein Structure
-
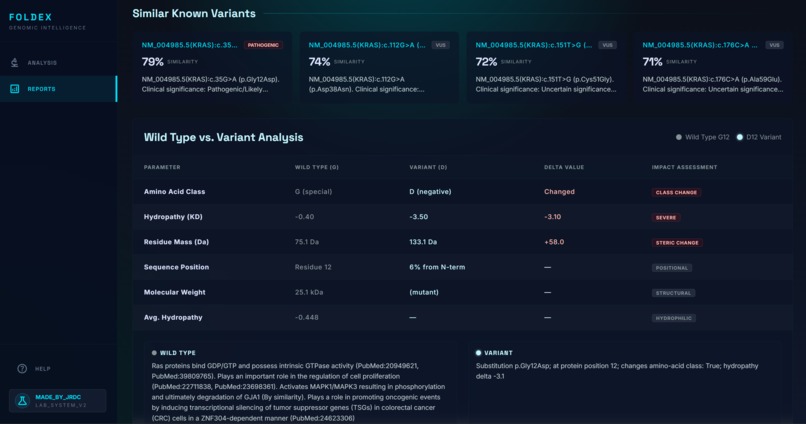
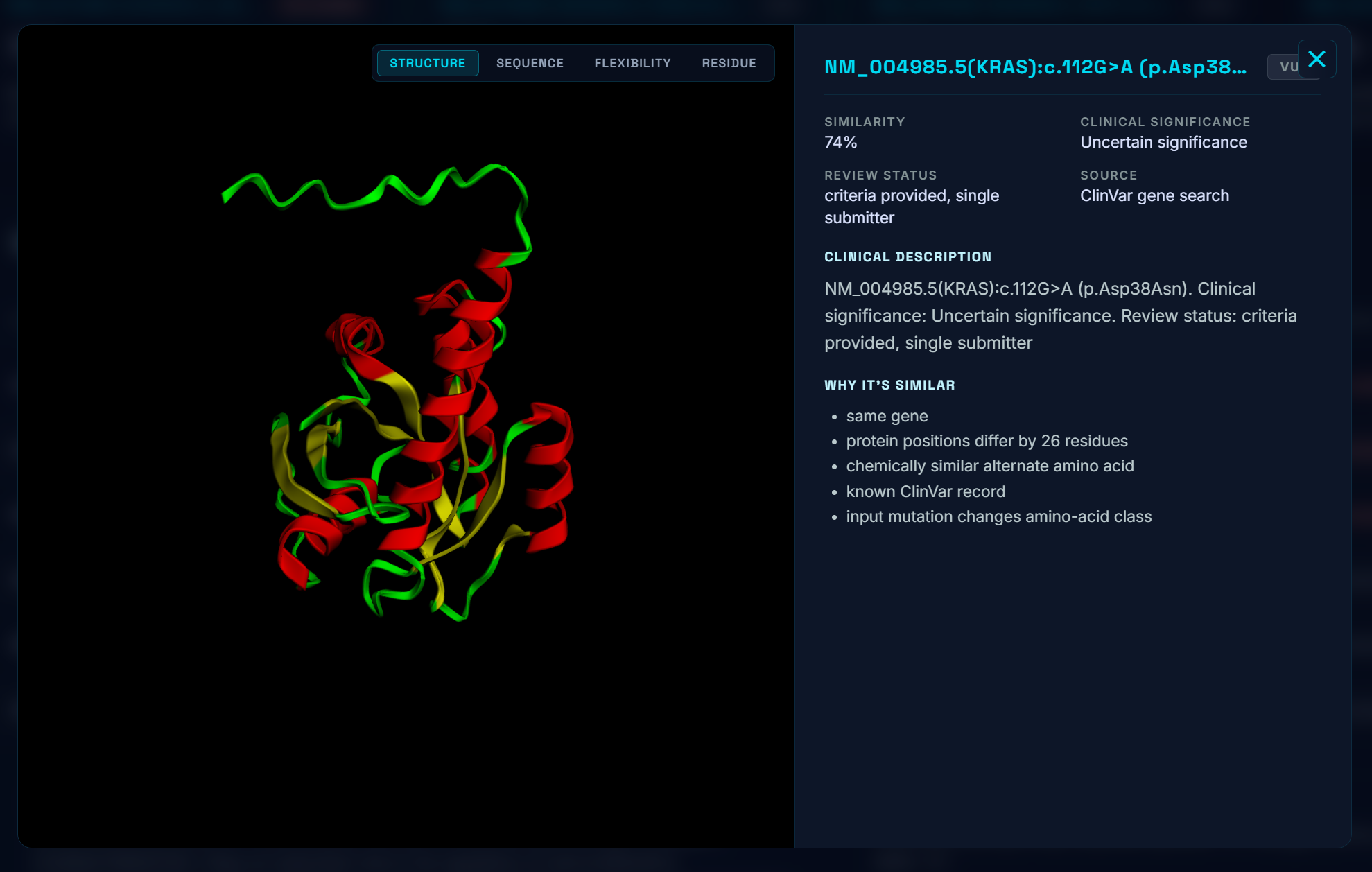
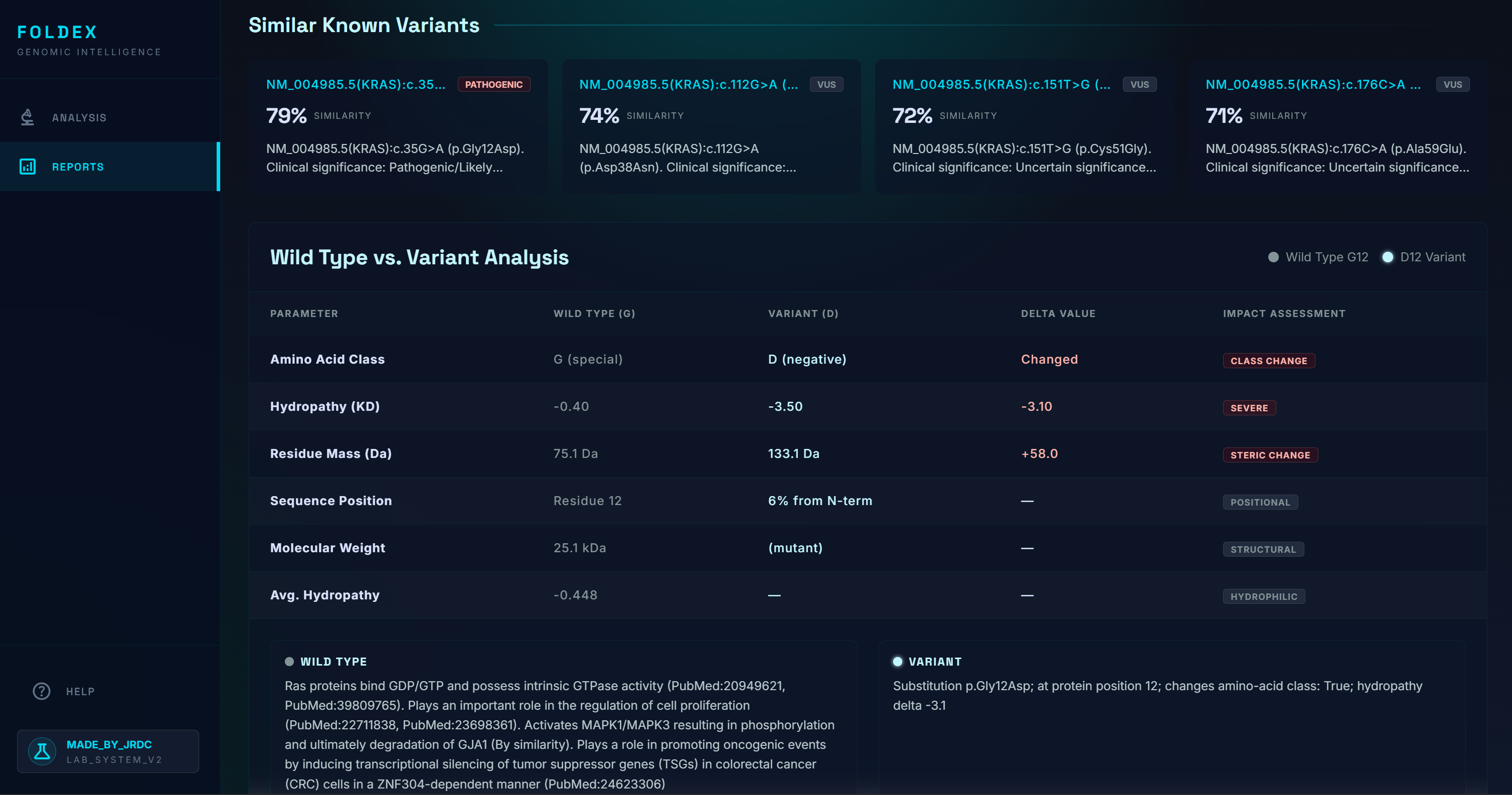
Similar Proteins
For live demo tests, example text inputs include: "KRAS p.Gly12Asp", "GJB2 p.Val27Ile" (last 3 is capital i, then lowercase l, then lowercase e), "MTHFR p.Ala222Val"
Inspiration
A "variant of uncertain significance," or VUS, is what a lab calls a mutation it has found in a patient's DNA but cannot yet classify as harmful or harmless. For the patient, that uncertainty is often more distressing than a clear diagnosis would be. Inherited cancer risk, rare childhood diseases, and unexplained neurological symptoms all hinge on the interpretation of these genetic grey areas. To provide clarity, we are currently synthesizing findings from the latest medical literature and querying specialized genomic databases for known similar variants. By analyzing this specific VUS alongside similar variants, we aim to uncover the evidence needed to transform an ambiguous lab result into a definitive clinical insight.
The scale of the problem is striking. A 2023 JAMA Network Open study of 1.7 million patients found that 41% had at least one VUS in their genetic testing report, and that only 7.3% of unique VUSs were reclassified across the study's eight-year window. The standard interpretation workflow requires a researcher or clinical geneticist to manually cross-reference half a dozen databases, retrieve the relevant protein structures, search for similar mutations in the literature, and write a synthesized report. Per variant, this can take days to weeks. The cost is so high that most rare-disease cases never receive the careful workup they deserve.
In Machines of Loving Grace, Dario Amodei describes a future in which AI compresses decades of biomedical progress into years. We wanted to build a small piece of that future: a tool that automates the evidence-gathering and structural-comparison work, and uses Claude to assemble the findings into a transparent report a clinician or researcher can actually use. Not as a diagnosis. As a head start.
What it does
FoldEx accepts a single gene variant and produces a complete research-support report in under two minutes.
Today, a clinical geneticist preparing a variant interpretation typically opens eight browser tabs, queries five separate databases by hand, downloads protein structure predictions, runs comparison software, reviews the literature, and writes a Word document. The work takes days.
With FoldEx, the same workflow produces:
- A plain-language summary of the mutation and its location within the protein
- A pathogenicity prediction with the underlying scores cited
- Population frequency data showing how common the mutation is across ancestries
- An interactive 3D model of both the wild-type and mutated protein, with the changed amino acid highlighted
- The five to ten most biologically similar mutations that have been studied, ranked by domain, residue distance, amino-acid property change, and pathogenicity-score similarity. We specifically scan through the most up to date research on bioRxiv, NCBI, etc for these known variants.
- A structured final report that clearly separates established clinical evidence from computational prediction Every claim in the report links back to a specific record in a real bioinformatics database. There are no invented variants and no fabricated citations.
FoldEx is designed for genetics researchers investigating novel variants, clinical geneticists preparing variant interpretations, rare-disease advocates and small clinics that lack a full bioinformatics team, and educators and students learning how variant interpretation works.
How we built it
Once the user submits a variant, Claude parses the input into standard HGVS notation, which is then validated against bioinformatics tools (VariantValidator and Ensembl VEP). The validated variant is sent in parallel to AlphaMissense via Ensembl VEP for pathogenicity scoring, ClinVar for clinical assertions, gnomAD for population frequency, and UniProt for protein metadata. The wild-type protein structure is retrieved from AlphaFold DB; the mutant structure is predicted by ESMFold. Candidate similar variants are pulled from the same gene, domain, and neighboring residues, then ranked. Finally, Claude synthesizes the structured evidence into a final report.
The frontend is built with Vite, React, and TypeScript, with Mol* providing the interactive 3D viewer. The backend is FastAPI with a Redis-backed job queue. Slow steps such as ESMFold structure prediction run as background workers so the UI can show partial results immediately.
The most consequential design decision was treating Claude as a reasoner over evidence, never as a source of evidence. Claude can rank candidate variants we provide, explain why two variants are biologically similar, and write up findings — but it cannot invent a variant, fabricate a citation, or make a claim we have not already pulled from a real database. We enforce this with strict JSON schemas: every claim must declare an evidence field pointing to a specific pipeline record and a limitations field flagging what is uncertain. Outputs that reference unfetched sources are rejected by a validator and retried.
Challenges we ran into
Lab reports are inconsistent. Different labs use different notations for the same mutation, and a single report often contains the variant in multiple formats. We layered Claude's extraction on top of bioinformatics validators so that ambiguous inputs are rejected rather than silently guessed.
Mitigating model overconfidence proved more complex than anticipated, as LLMs naturally tend toward authoritative prose even with insufficient evidence. We addressed this by implementing a custom quantitative similarity score to anchor the model’s reasoning. By providing Claude with specific annotations and feature-extraction data, we prompted it to synthesize an explanation based strictly on those parameters rather than general inference.
Finally, AlphaFold DB only stores wild-type structures. To produce a meaningful structural comparison, we had to combine AlphaFold (wild-type) with ESMFold (mutant), align the two structures, and highlight the residue that differs.
Accomplishments that we're proud of
Every sentence in the final report is traceable to a specific record in ClinVar, gnomAD, Ensembl VEP, or UniProt. This kind of traceability is unusual for LLM-generated medical content and was non-negotiable for us.
The end-to-end demo works: a user can paste a real-world variant and receive a structurally-grounded report in under two minutes. Ethical guardrails are integrated rather than appended: disclaimers appear in the UI, in the report header, and in the JSON output. The Claude prompt design actively resists overconfidence by requiring weak or conflicting evidence to be flagged rather than hidden.
What we learned
The gap between "AI that sounds plausible" and "AI that is safe to deploy near a clinical workflow" is mostly a question of constraining inputs and requiring structured, auditable output. It is not primarily a question of model size or fine-tuning. The genuine value of a project like FoldEx lies in integration, not in any single API or model. Bioinformatics tooling is rich but fragmented; a thoughtful glue layer is most of the win.
Additionally, designing for a specific real user, a geneticist who will be held professionally accountable for the interpretation, produces a much sharper product than designing for "users in general." Many of our hardest design decisions resolved themselves once we asked what that specific user actually needed to trust the output.
What's next for FoldEx
We plan to add OCR-based ingestion for scanned lab-report PDFs, integrate literature evidence from PubMed and LitVar alongside database evidence, and incorporate experimental data from MaveDB. A batch mode for research labs analyzing dozens of variants at once is in design, as is a researcher export that produces an evidence packet ready for inclusion in a manuscript.
For FoldEx to reach the people who need it most, it has to survive beyond the MVP. Our sustainability plan is a cross-subsidy model: pharma R&D and large commercial labs pay for the tool because they have the budget and a commercial use case, academic medical centers pay reduced rates in exchange for validation work, and rare-disease foundations and under-resourced clinics receive free or near-free access. The global genetic testing market was estimated at $14.5 billion in 2025 (Straits Research; corroborated by Grand View, 360iResearch, and Market Data Forecast), and is projected to grow at a 10–20% CAGR depending on methodology. There is ample room for a sustainable layer focused on accessible variant interpretation. Mission-driven organizations like Khan Academy, Signal, and Mozilla have shown that this kind of mixed-funding model can be durable.
Built With
- claude-api
- docker
- fastapi
- python
- react
- tailwindcss
- typescript
- vite


Log in or sign up for Devpost to join the conversation.