-
-

-
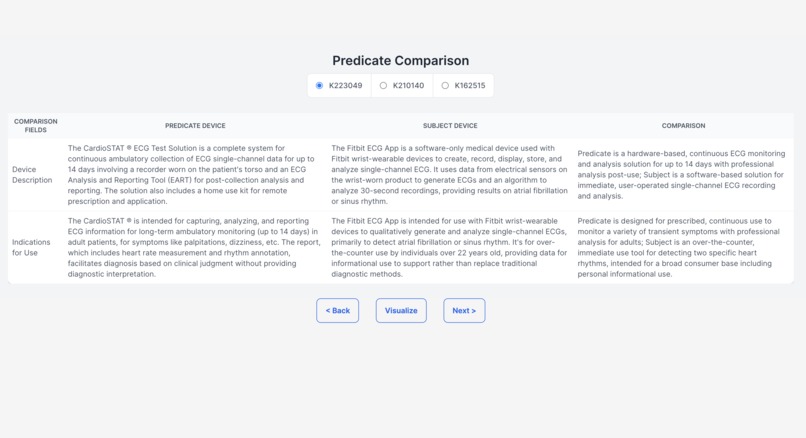
Generating comparison table of predicate devices (part 1)
-
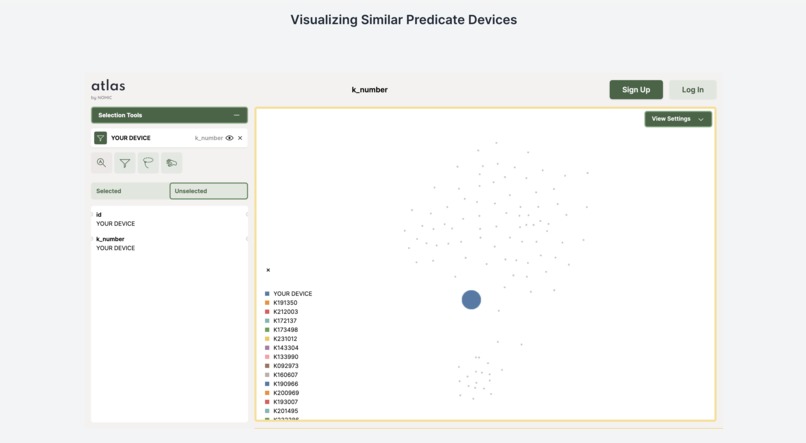
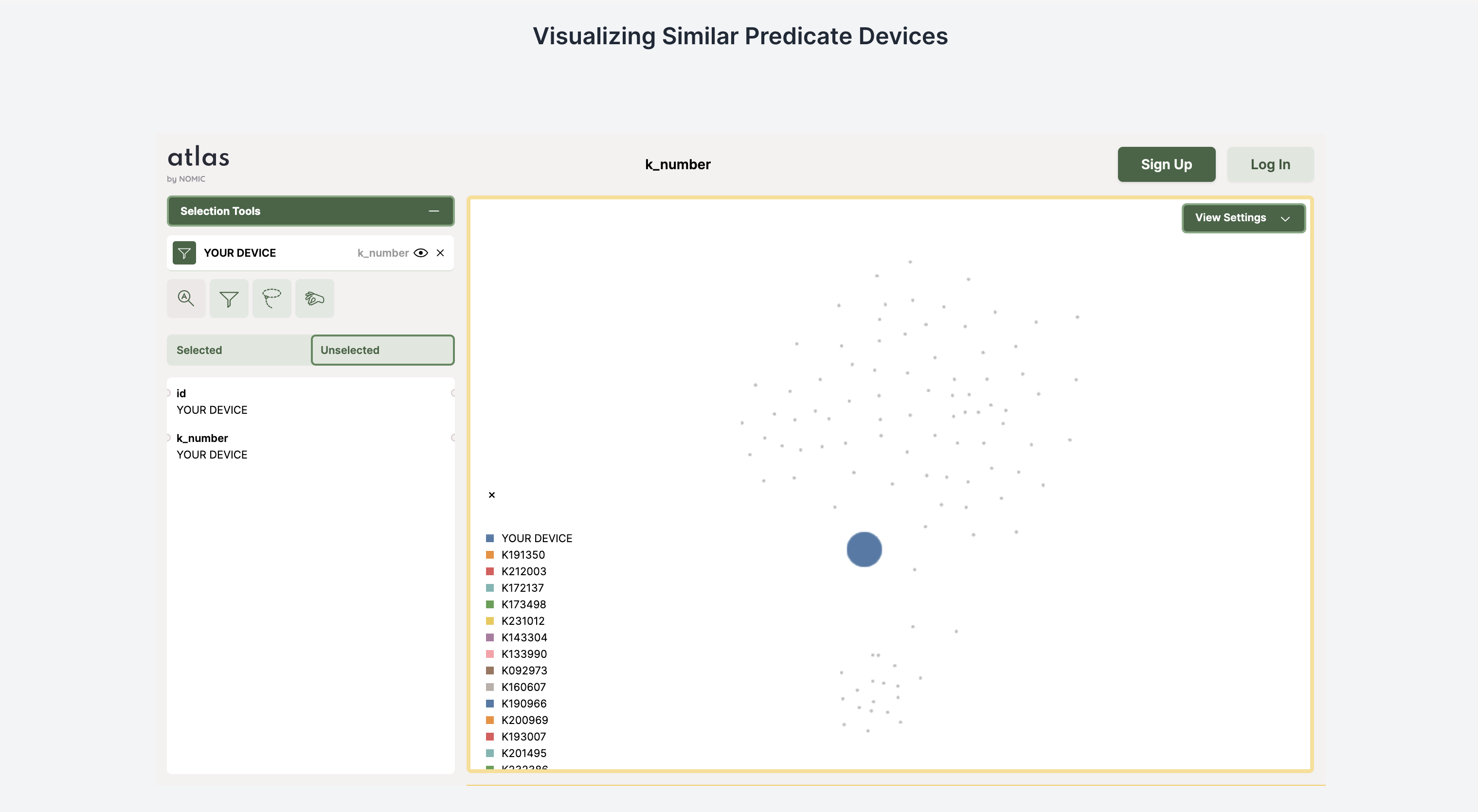
Visualization of similar devices
-
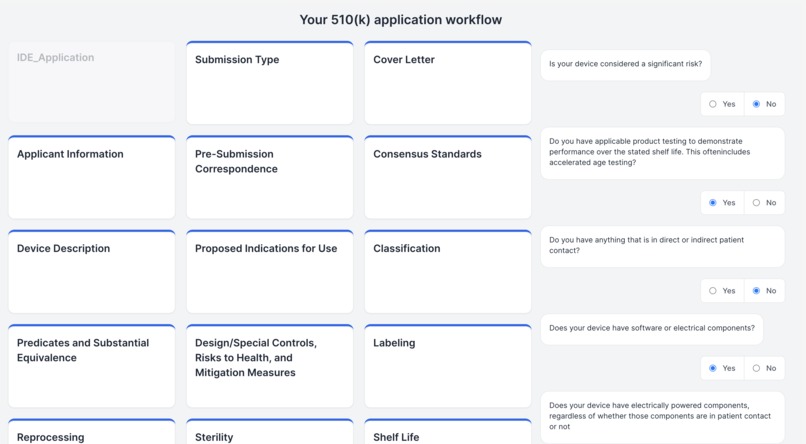
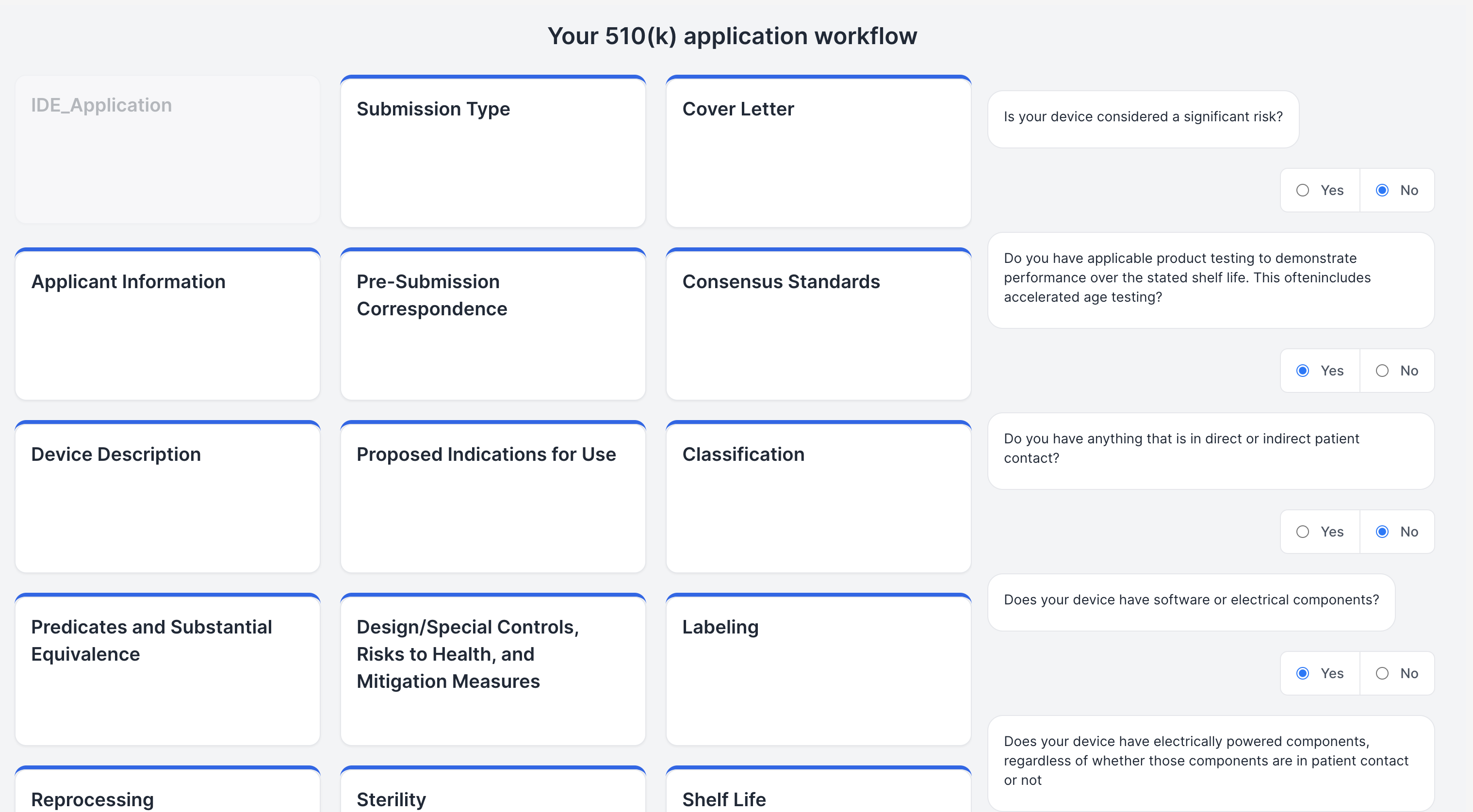
Personalized Regulatory Pathway
-
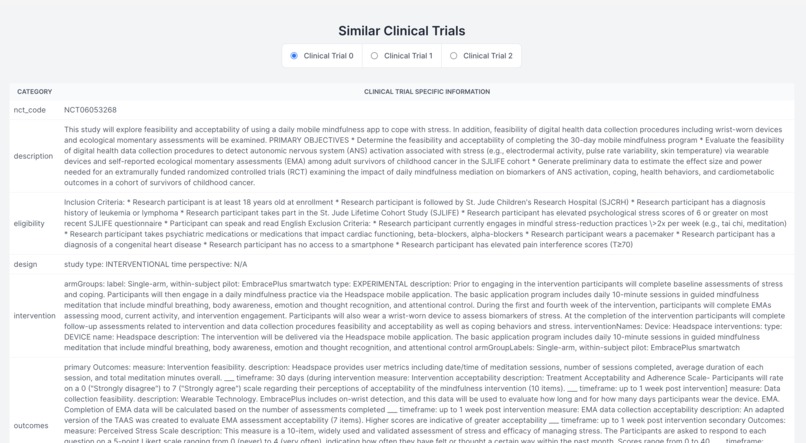
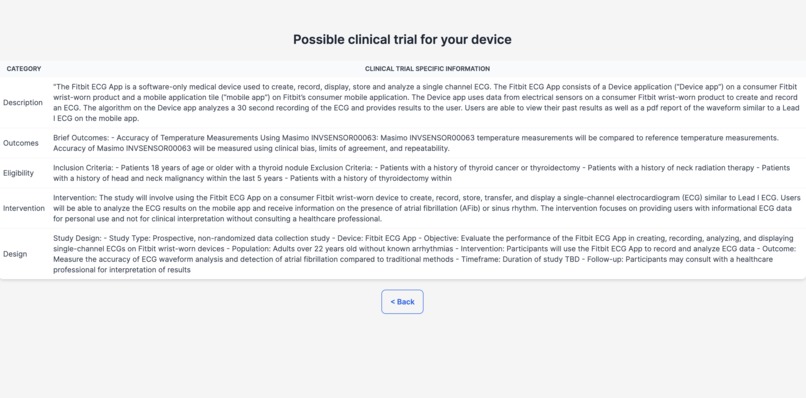
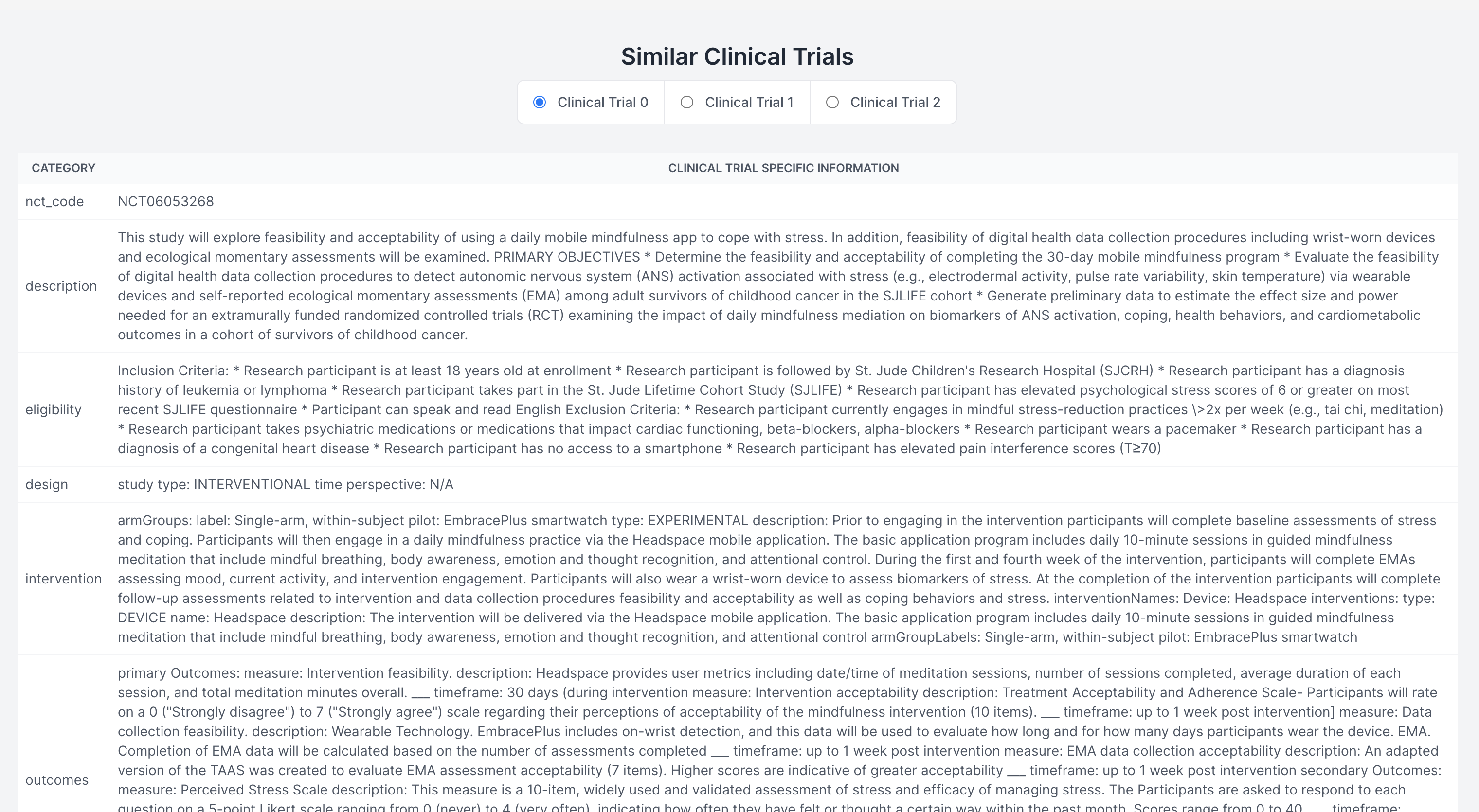
Finding Similar Clinical Trials
-
Clinical Trial Generation


Inspiration
Medical device companies spend on average between $20-40K on FDA consulting services and around 9 months to complete just the 510k approval process which allows you to legally market your device. With the rise of software enabled devices (such as wearables etc), the development time for these devices are decreasing while regulatory timelines haven’t changed, hindering and even discouraging startups from going through the FDA process. Our team of 3 MIT graduate students have previously developed wearable medical devices and spent months designing study protocols, revising IRB applications, and doing literature review before even starting pilot testing of the device.
After numerous conversations with other medical device entrepreneurs and FDA consulting services, we uncovered widespread frustrations with the FDA's overwhelming, non-intuitive, and scattered information landscape, particularly for medical devices. Driven by these challenges, we developed FastTrackFDA—a transformative platform engineered to streamline the FDA journey starting from device conception and guiding development until FDA clearance.
What it does
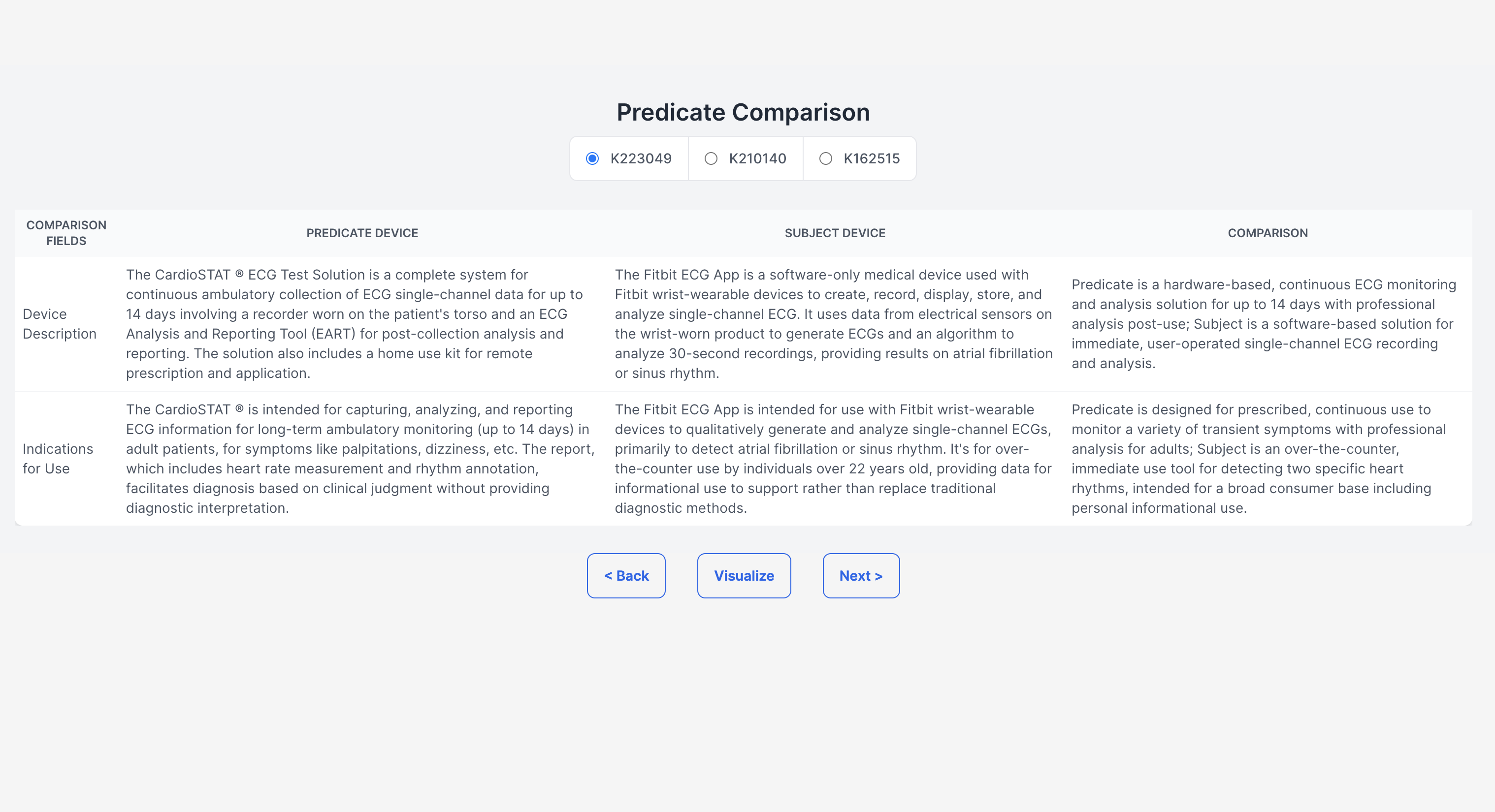
Demystifying the FDA journey, our platform empowers medical device startups and companies by guiding them seamlessly through the approval process. It aids in identifying predicate devices and crafting clinical trial protocols, significantly reducing the reliance on consultants. The FDA's website, often criticized for its cluttered guidance documents, constant revisions, voluminous and complex databases, and lack of personalization, poses a significant challenge, especially for medical device manufacturers. Our solution is an intuitive user interface, crafted with input from device developers, designed to streamline the description and intended use case of your device. It enables a comprehensive understanding of all market entry requirements, facilitating the creation of predicate devices and clinical trial designs. This approach not only accelerates the approval process and reduces costs but also diminishes the dependence on external consulting services. (1) Finding Substantially Equivalent Devices: Identifying a substantially equivalent device is crucial in determining the appropriate regulatory pathway for your device. For FDA 510(k) approval, it's imperative that your device is matched with a substantially equivalent 'predicate' device. Traditionally, consultants might spend 20-40 hours on this task alone. Our platform leverages vector search and matrix similarities, to pinpoint the most compatible predicate devices, using a semantic comparison with your device. (2) Personalizing the Regulatory Workflow: With the FDA's mandate from October 2023 requiring all 510(k) submissions to follow an e-submission template, our platform standardizes yet personalizes the steps necessary for compliance. (3) Generating Clinical Trial Designs: To streamline the development of study designs, our platform displays clinical trials for devices that are semantically similar to yours, sourced from the clinicaltrials.gov database. We then generate potential designs, incorporating inclusion/exclusion criteria, intervention modules (control and experimental groups), study procedures, and outcome measures, based on a comprehensive analysis of clinical trial data. This refined approach not only clarifies the path to FDA approval but also positions our platform as a pivotal tool in bringing medical devices to market more efficiently and cost-effectively.
How we built it
Our application consists of a Next.Js frontend and Flask backend. For generating our dataset of all the 510k summaries, we developed our own custom pdf extractor to extract different sections and tables from the 510K summary documents on the FDA site. In order to find the best predicate device we developed a matrix similarity algorithm using the vector similarity search built off of our data storage in Pinecone. Then we used openai GPT-4-turbo to generate comparison tables among the two devices which is a section of the 510k document. To visualize the database (in order for users to see where their device lies in the space of all similar devices), we used Nomic’s Atlas module. Lastly, we automatically collected a dataset of clinical trial designs from clinicaltrials.gov and used Together API to finetune a collection of clinical trials generation models and also used openAI’s LLM for trial generation.
Challenges we ran into
Table extraction: Each PDF document for 510k summaries has a different format (some are scanned copies and some are pdfs with differently formatted tables and various headers that aren’t standard). We tried multiple approaches using computer vision, ocr, table extraction libraries, and heuristics and ultimately creating our own custom approach that ran faster than the other approaches in order to populate our database with all of the previous records of 510ks. Deployment: Due to some dependencies, we had an issue with deploying the full application so we could deploy the frontend and backend separately.
Accomplishments that we're proud of
We’re really proud of actually solving a use case that we’ve seen a huge need for first hand and no real solution. We truly believe that cost, time, or ambiguity should not be the reason that a medical device which can transform a person’s life doesn’t get to market soon enough.
What we learned
- Prioritization of features and shipping an MVP that addresses the most impactful ones
- Table extraction among pdfs isn’t a solved task and there are many nuances
- The FDA process is very complex
What's next for FastTrackFDA
Our overall mission is to be a central platform for all device companies to start using when they begin conception of a device.
Future steps that we plan to build out soon are:
- Expanding to other areas of the FDA process such as quality Assurance: 75% of the time that a consultant spends is revising existing drafted material and making sure it abides by the Refusal to Accept checklist documentation and guidance documents for each section. We plan to user our table extraction tool to extract this RTA document and do automatic checking before the person creates the table.
- Improve table extraction to be more generalizable: We hope users can ask interactive questions about guidance documents but only once they’ve been parsed perfectly.
- Continuous learning and multi-agent reviewal- Since newer FDA device approvals are most important, we plan to integrate a continuous learning approach that constantly updates the data as soon as a new device is approved.
- Multi-agent reviewal: For each part of this workflow, we hope to embed more specific knowledge about the decision making process of a FDA consultant. For example, for determining clinical trial design we hope to embed an “clinical trial expert” agent that checks whether the trial’s inclusion/ exclusion reduces bias, its trial design is safe, and potential suggestions.
Built With
- flask
- next.js
- openai
- pinecone
- sentence-transformers
- tailwind
- together

Log in or sign up for Devpost to join the conversation.