


Inspiration
Drug discovery is slow, expensive, and deeply fragmented. Researchers working on early-stage candidate identification spend significant time manually moving data between disconnected tools — a scoring model here, an ADMET calculator there, a knowledge base somewhere else — with no unified way to query or reason over the results. I kept running into this problem when studying computational drug discovery. There was no single open-source system that could take a target protein and give you ranked, drug-like candidates with similarity to known drugs, ADMET properties, and a knowledge graph — all in one automated pipeline. So I built one.
What It Does
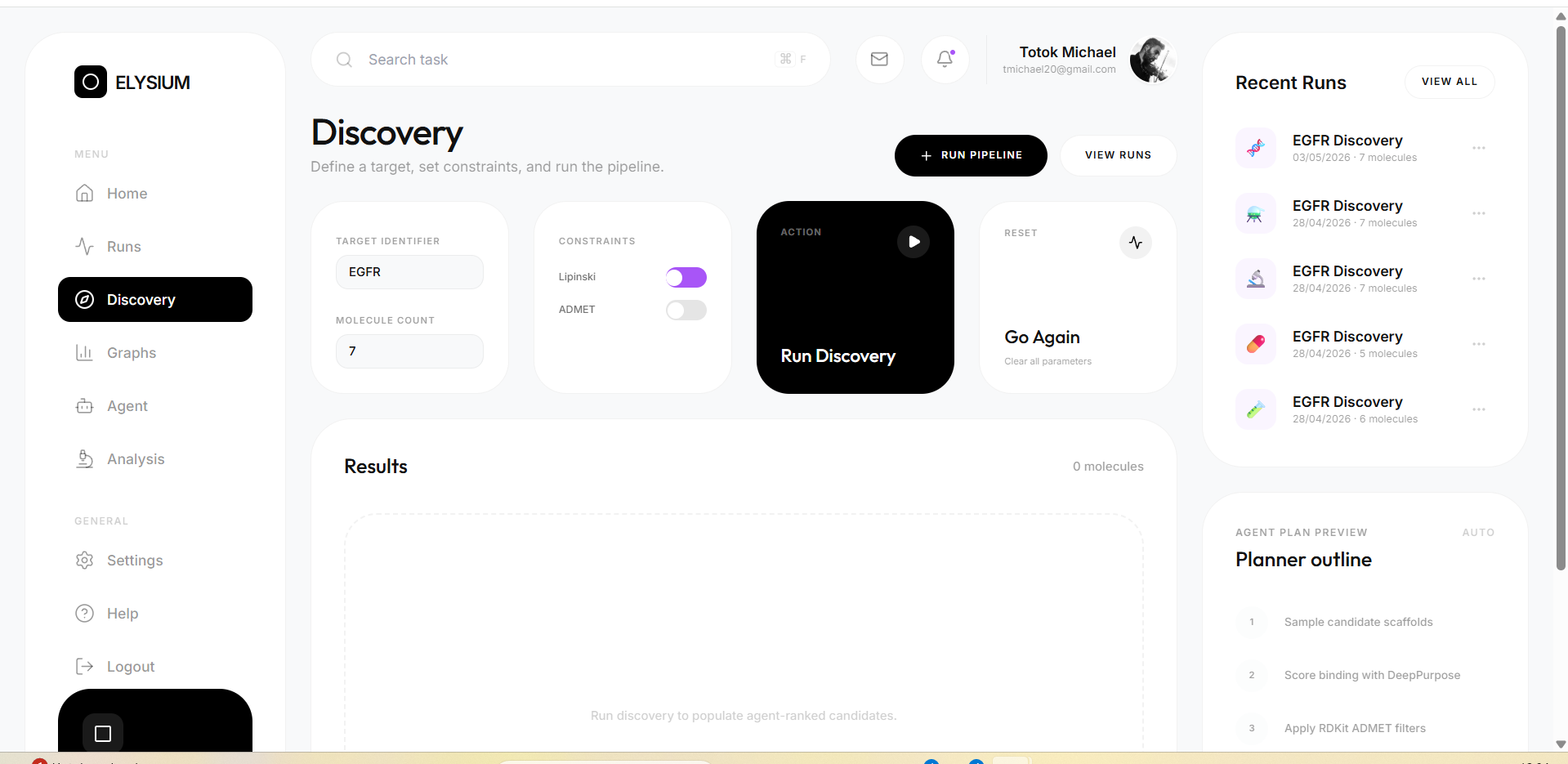
ELYSIUM is a full-stack AI drug discovery platform that automates the early-stage pipeline from target to ranked candidates.
You type a target protein. ELYSIUM does the rest.
Here is what happens in the background in under 12 seconds:
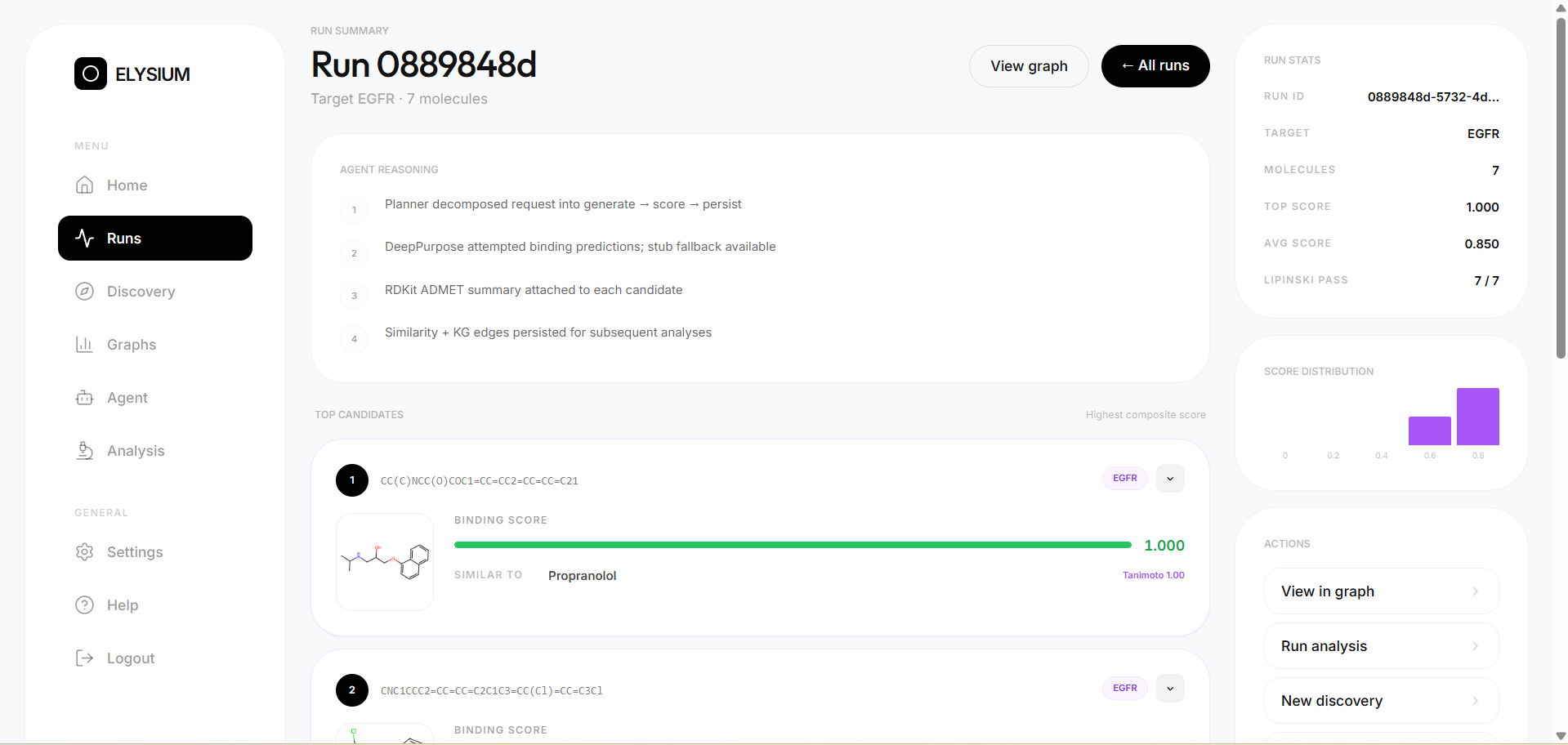
- Candidate generation — molecules are sampled from a curated drug-like SMILES library spanning 12 therapeutic classes
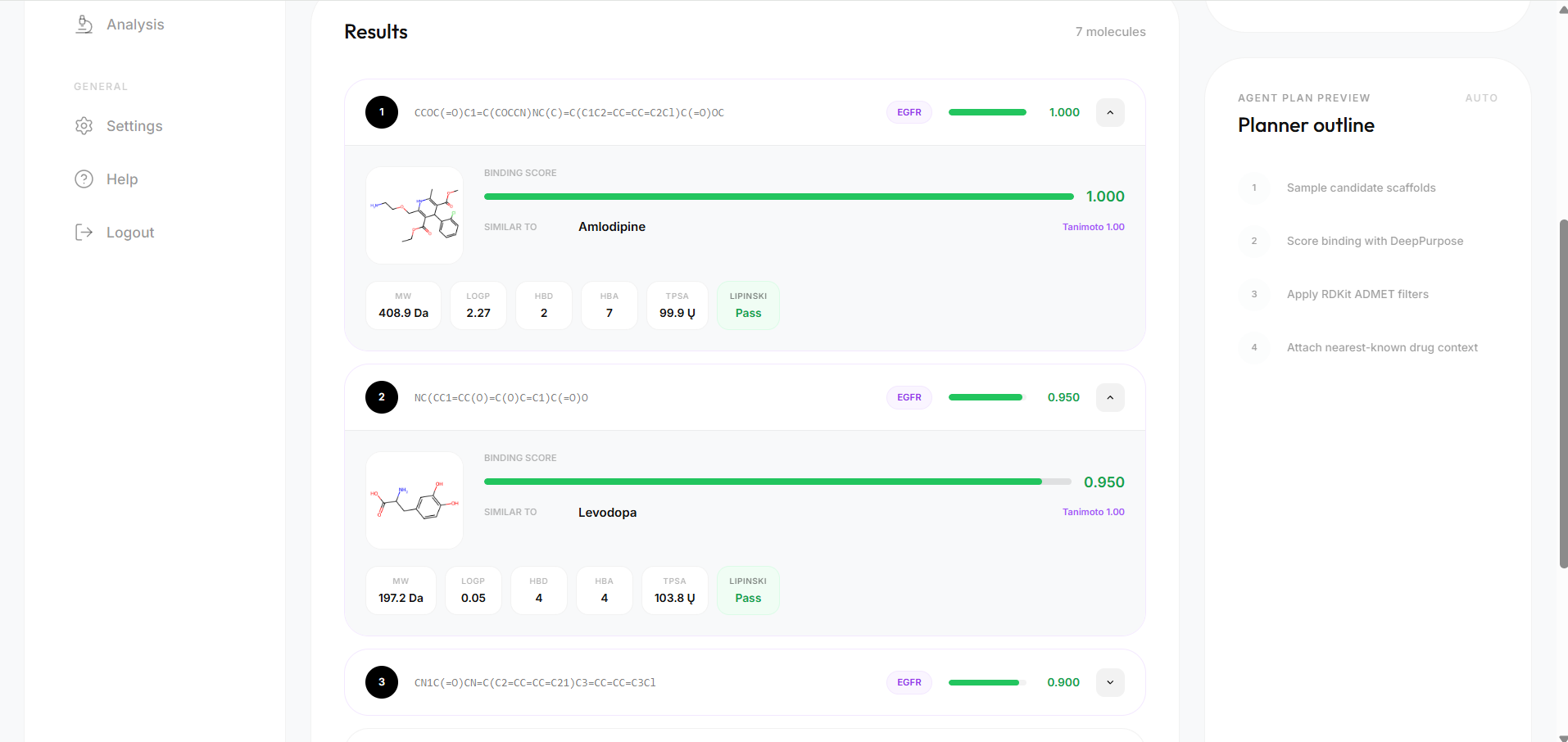
- DTI scoring — DeepPurpose predicts binding affinity for each candidate against the target, normalised to [0, 1]
- ADMET profiling — RDKit computes molecular weight, LogP, TPSA, HBD, HBA, and Lipinski violations for every molecule
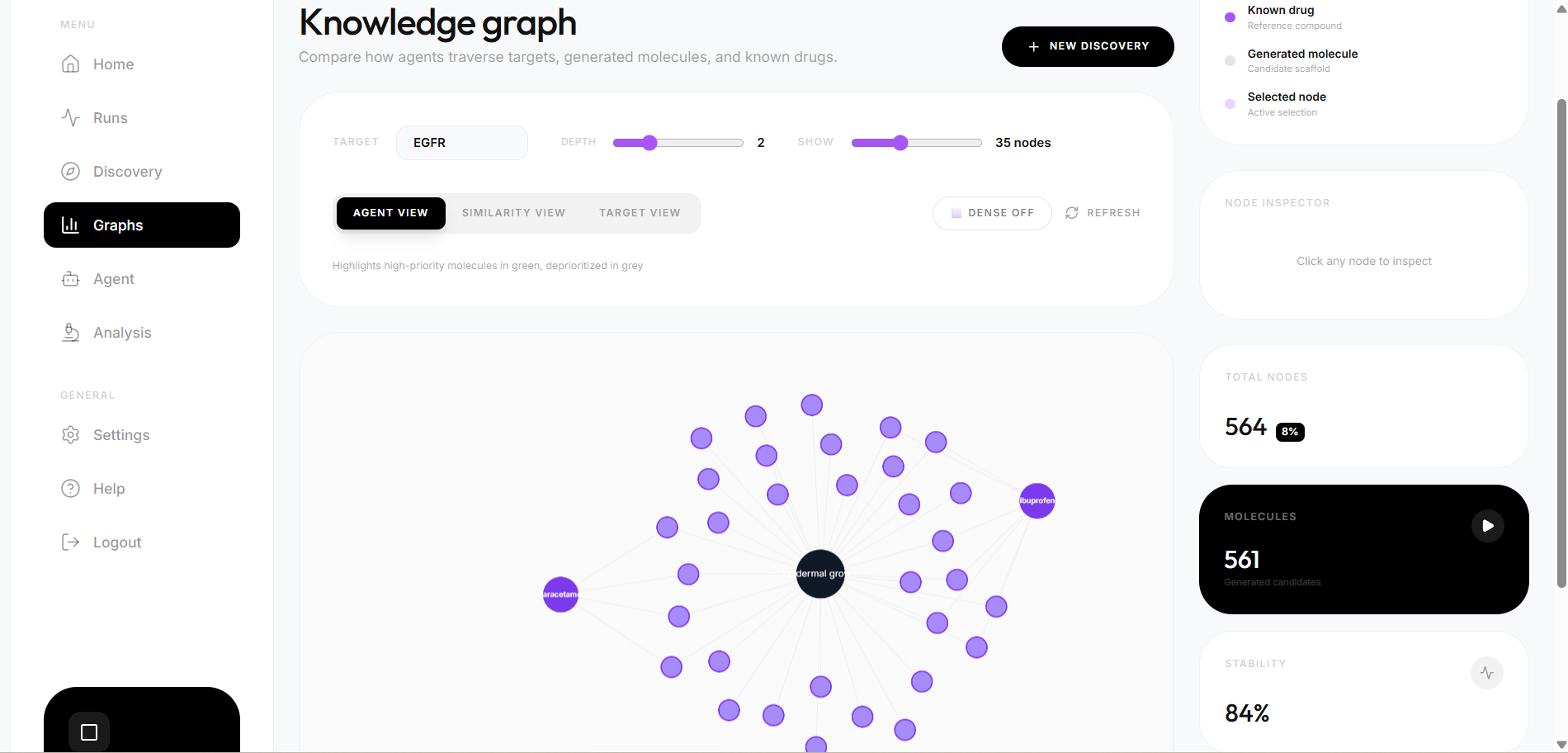
- Dual similarity search — Morgan fingerprint Tanimoto similarity AND ChemBERTa semantic cosine similarity both run in parallel, giving two independent hypotheses about which known drug each candidate resembles most
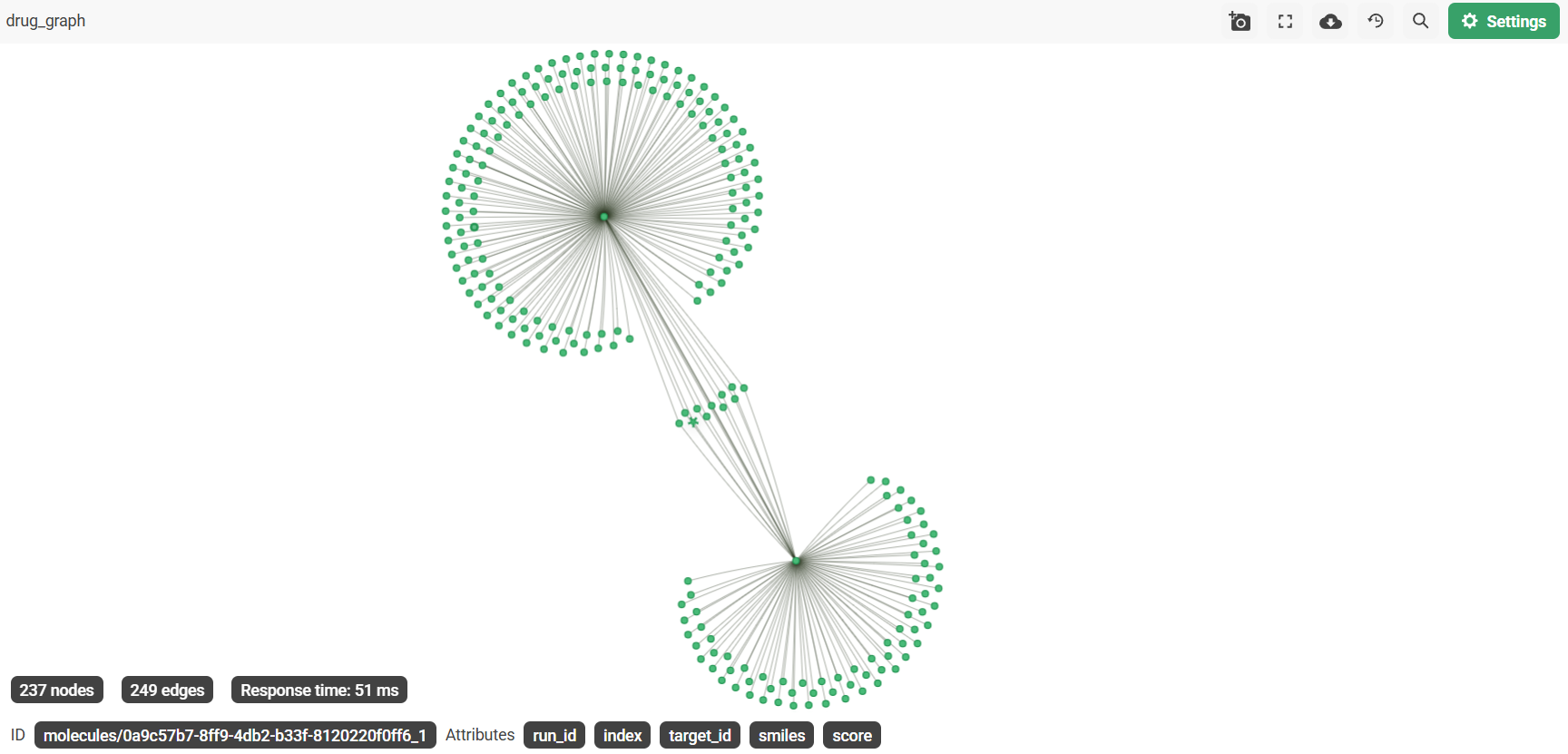
- Knowledge graph persistence — every molecule, target, and drug similarity edge is written to both SQLite and ArangoDB, enabling multi-hop graph traversal across runs
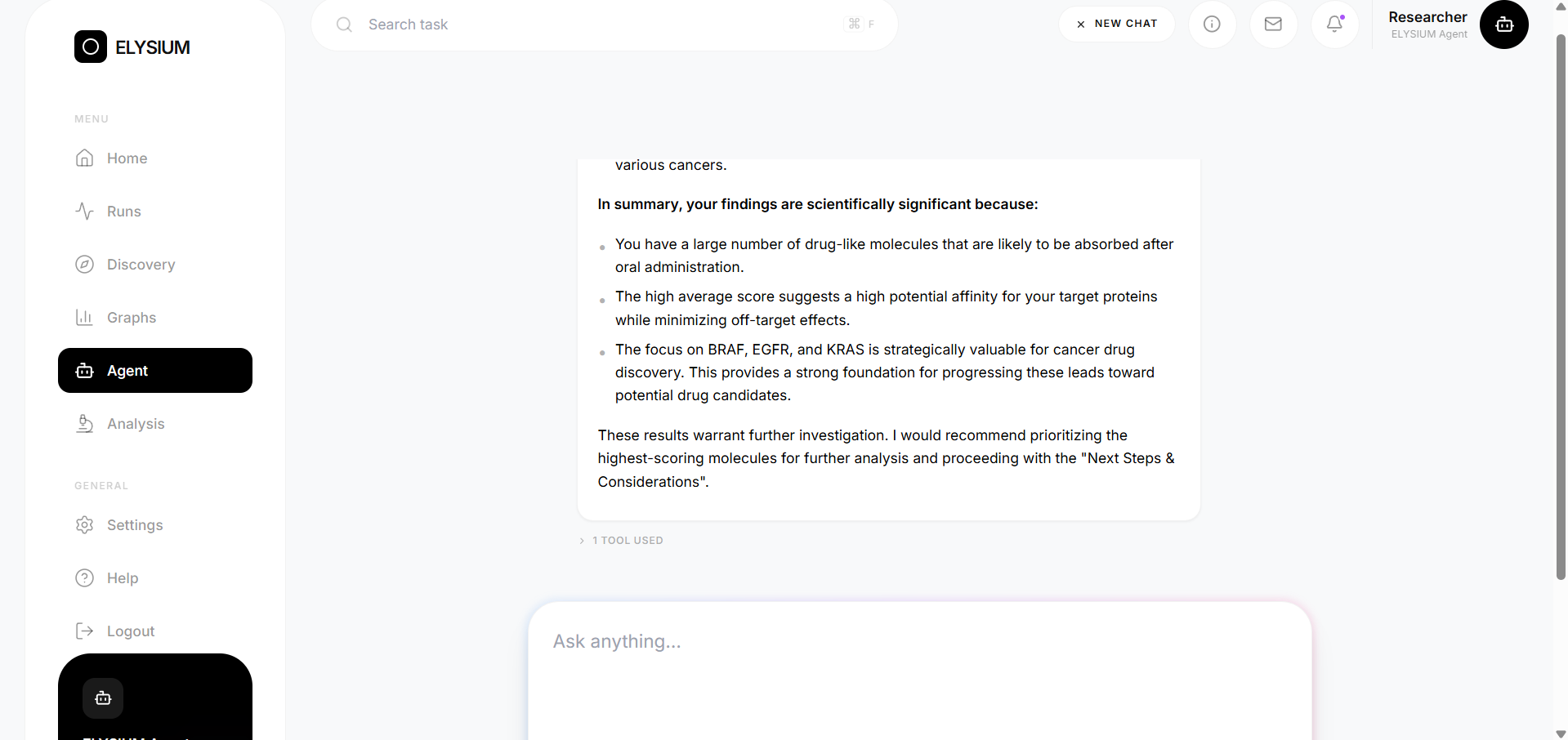
- Natural language querying — a ReAct-pattern LLM agent lets you ask plain-English questions about your results and get grounded answers from real database values, not hallucinations
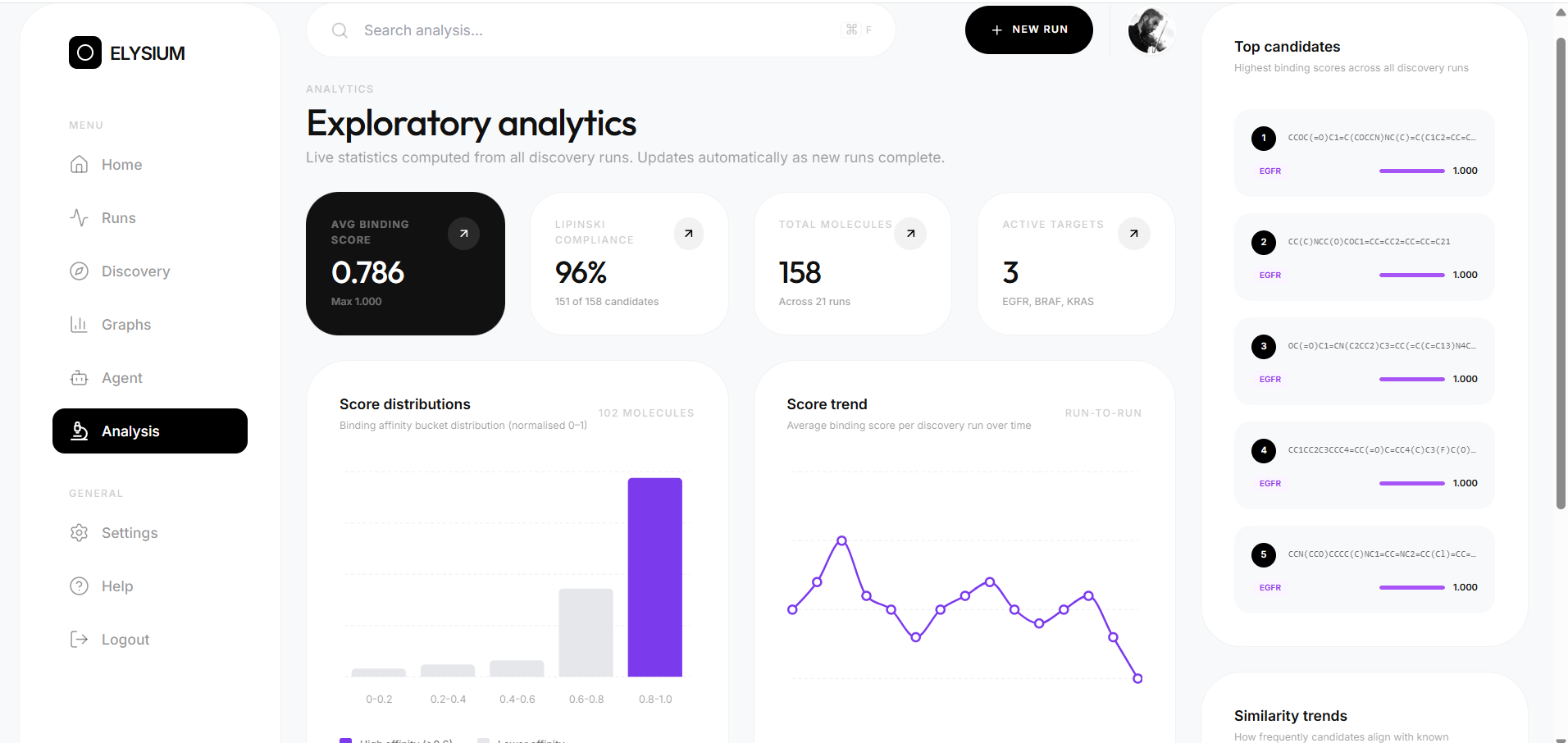
Experimental results across EGFR, BRAF, and KRAS:
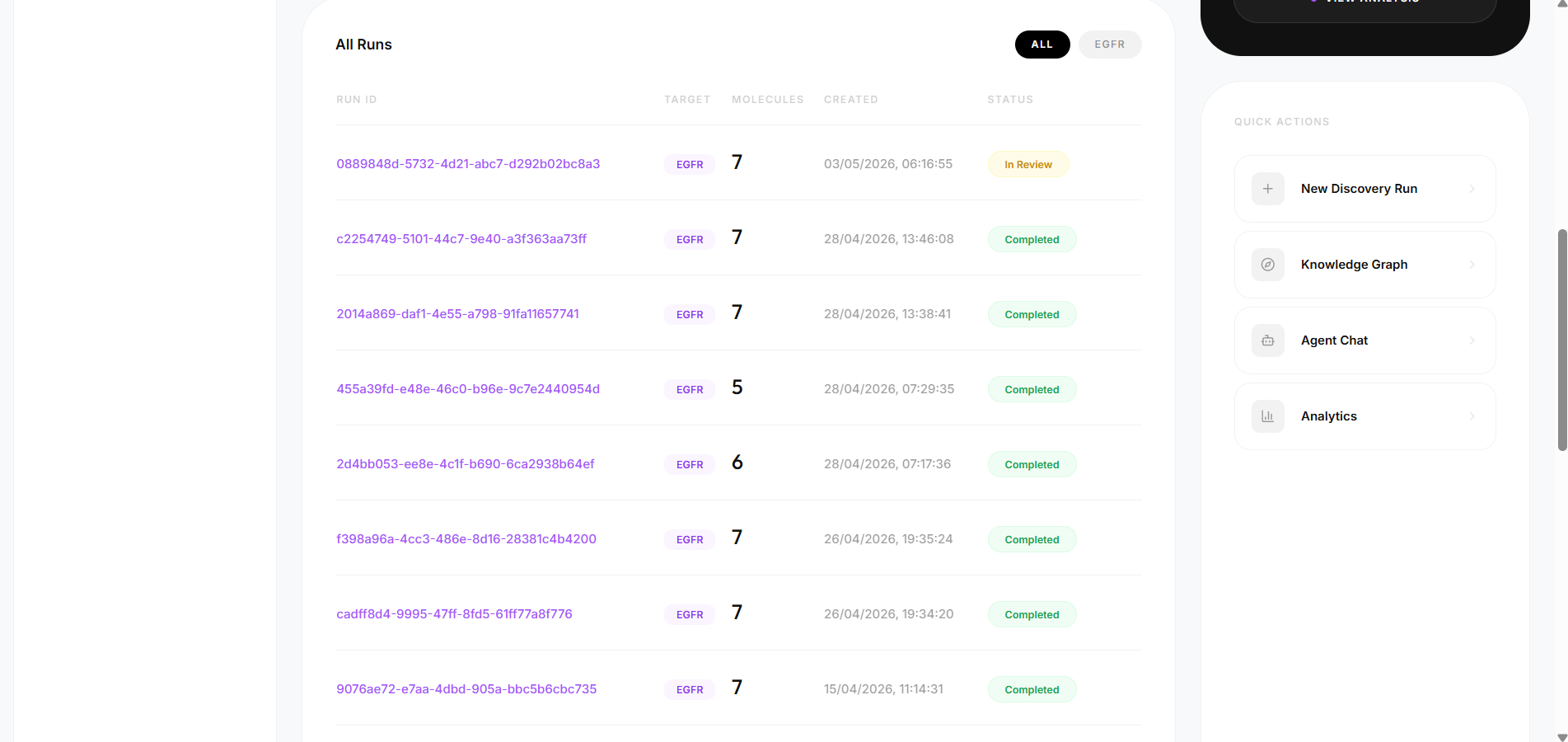
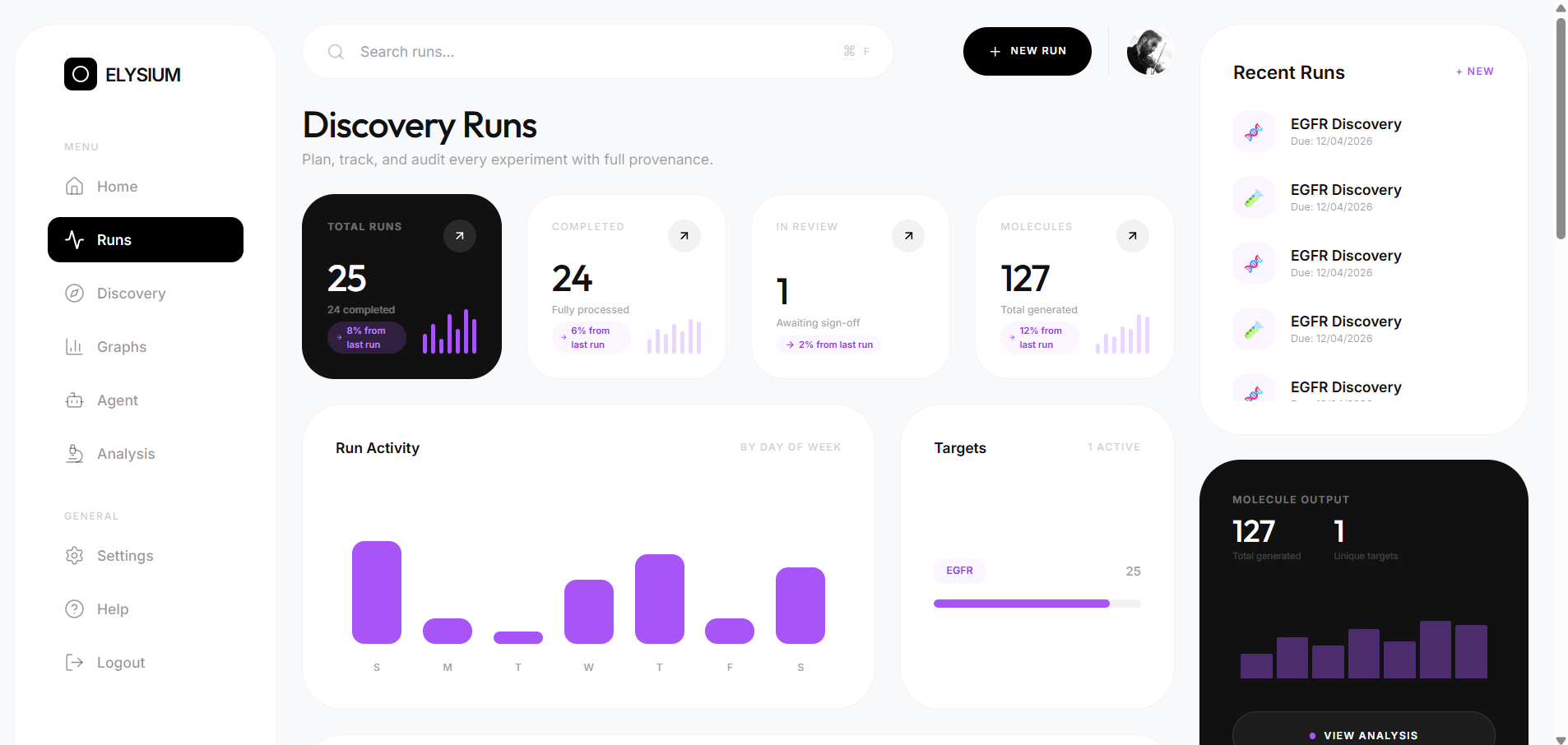
- 83 candidate molecules generated across 9 discovery runs
- 90.7% overall Lipinski rule-of-five compliance rate
- 0.775 average predicted binding score
- 95.3% Lipinski compliance for EGFR candidates
- Agent correctly resolved multi-turn queries including anaphoric references across conversation turns
The platform is fully accessible through a React frontend — no programming knowledge required to run a discovery pipeline.
Connection to SDG 3 — Good Health and Well-Being
SDG 3 calls for ensuring healthy lives and promoting well-being for all. One of its targets specifically supports research and development of medicines and access to safe, effective treatments.
ELYSIUM contributes to this by lowering the barrier to early-stage drug discovery research. Most computational tools in this space require significant programming expertise, expensive hardware, or proprietary licenses. ELYSIUM runs entirely on open-source components, works on consumer hardware, and is accessible through a web interface — no programming knowledge required. A researcher at an institution without a dedicated computational chemistry team can run a complete candidate screening pipeline in seconds rather than days.
The system was validated against three well-studied kinase targets — EGFR, BRAF, and KRAS — chosen because they have known reference inhibitors, making them suitable benchmarks for evaluating pipeline correctness. The broader goal is a platform where researchers anywhere can run hypothesis-driven computational screening without needing industry-scale resources.
How I Built It
ELYSIUM is a three-tier system: a React/TypeScript/Tailwind frontend, a FastAPI Python backend, and a dual persistence layer combining SQLite with ArangoDB.
The scientific pipeline:
- DeepPurpose (DTI scoring) — pretrained CNN-RNN architecture on BindingDB, invoked via the DeepPurpose virtual screening API
- RDKit (ADMET profiling) —
Descriptors.MolWt,Crippen.MolLogP,rdMolDescriptors.CalcTPSA, Lipinski module for HBD/HBA - ChemBERTa (semantic embeddings) —
[CLS]token extraction fromseyonec/ChemBERTa-zinc-base-v1, normalised to unit vectors - FAISS (vector search) — indexed reference drug embeddings for efficient approximate nearest-neighbour retrieval
- ArangoDB (knowledge graph) —
BINDSandSIMILAR_TOedge collections in a named graphdrug_graphfor AQL traversal
Challenges
ChemBERTa cold-start latency. The model takes 15–20 seconds to load on first call. Solved by keeping it in memory across requests, but this needs proper pre-loading at startup in production.
OpenRouter rate limits. Free-tier LLM providers hit rate limits fast under testing load. Built a cascading fallback strategy that cycles through 10+ models automatically — this ended up making the agent more robust than originally planned.
Graph data when ArangoDB is unavailable. ArangoDB requires a running server, which isn't always guaranteed. Solved by wrapping all ArangoDB writes in per-operation error handlers so the core pipeline continues even if the graph database is down.
SMILES validity. The generation module initially produced invalid
SMILES for ~12% of attempts. Fixed by adding RDKit Chem.MolFromSmiles
validation at generation time and discarding invalid outputs before
scoring.
Making the UI actually usable. Getting a research-grade pipeline to feel accessible to non-programmers took more iteration than the pipeline itself. The Design system — Outfit font, purple accent, rounded cards, sidebar navigation — went through multiple complete rewrites before it felt right.
What I Learned
The science was the easy part. The hard part was making it work reliably as a system — handling cold starts, database unavailability, LLM rate limits, and SMILES validation failures all gracefully, without the whole pipeline crashing.
I also learned that dual similarity search (fingerprint + semantic) gives you genuinely different information. Tanimoto fingerprint neighbours like Aspirin suggest scaffold conservation. ChemBERTa semantic neighbours like Amlodipine — identified despite low structural overlap — point toward bioisosteric possibilities. Running both in parallel gives a medicinal chemist two independent directions for follow-up, which is something a single similarity metric can never do.
And honestly — I learned that the $2.6 billion / 15 year statistic isn't just a slide. It represents real people who don't get treatments in time. Building something that could meaningfully reduce that timeline, even at the early stage, felt like the most worthwhile thing I've done in three years of CS.
What's Next
Three concrete priorities:
1. Expand the molecule library. The current 50-molecule curated library is the biggest limitation. A filtered ChEMBL subset of 500–1000 Lipinski-compliant compounds would dramatically increase chemical diversity without needing a generative model.
2. Fine-tune the DTI scorer. DeepPurpose currently uses general pretrained weights. Fine-tuning on EGFR, BRAF, and KRAS bioactivity data from ChEMBL would give calibrated affinity estimates rather than general predictions — a necessary step before any experimental follow-up.
3. Native multi-hop agent reasoning. The current agent handles single-turn tool selection reliably. Exposing ArangoDB graph traversal as a native agent tool would enable complex multi-hop queries like "find all molecules that bind EGFR, are similar to an approved NSAID, and pass ADMET filtering" — closing the loop between graph-native storage and agentic reasoning.
Built With
- arangodb
- chemberta
- deeppurpose
- docker
- faiss
- fastapi
- huggingface
- openrouter
- python
- rdkit
- react
- sqlalchemy
- sqlite
- tailwindcss
- typescript
- vite

Log in or sign up for Devpost to join the conversation.