-
-
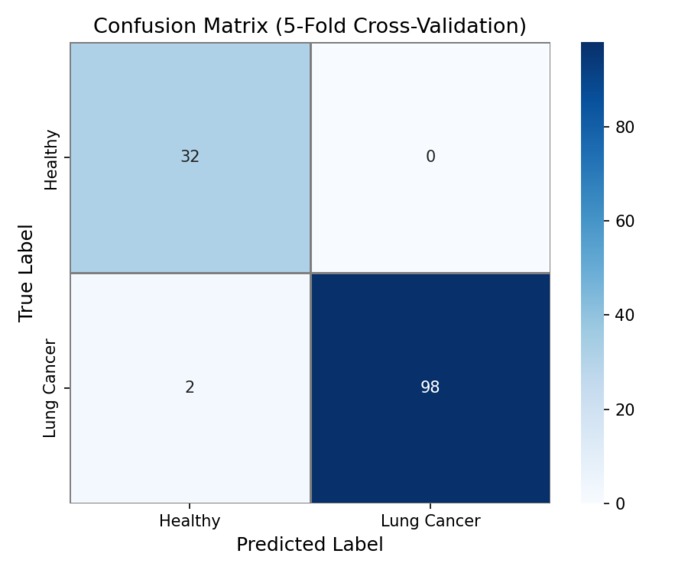
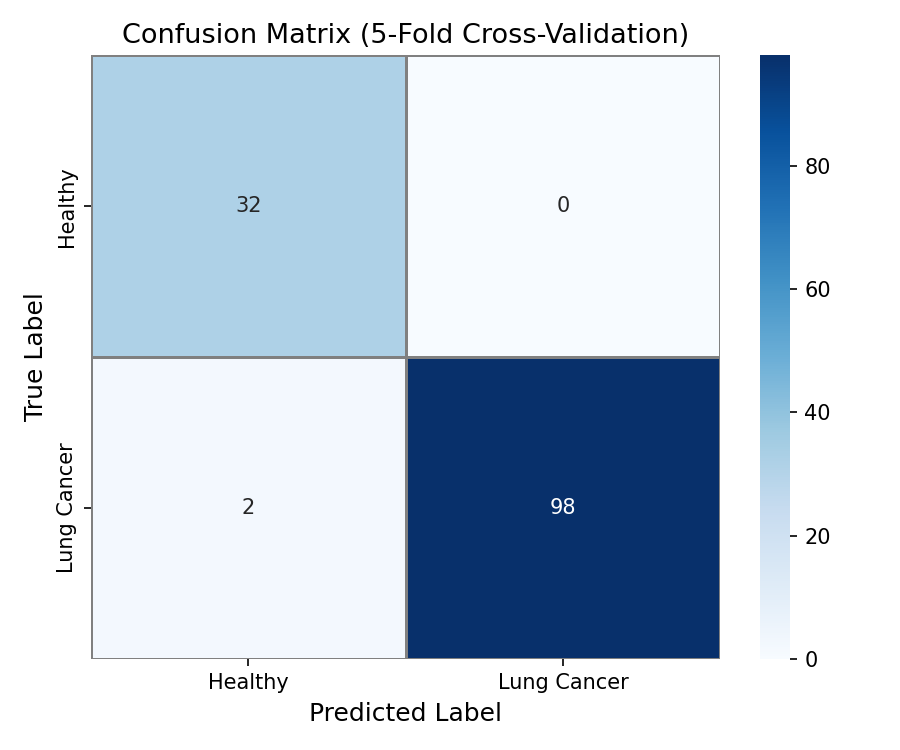
Confusion Matrix - (TP, FN, FP, TN) Pure numbers regarding the accuracy of the model
-
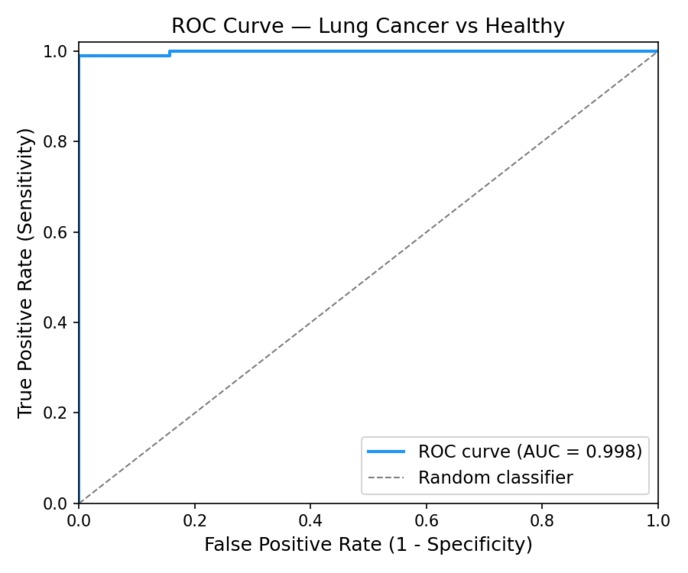
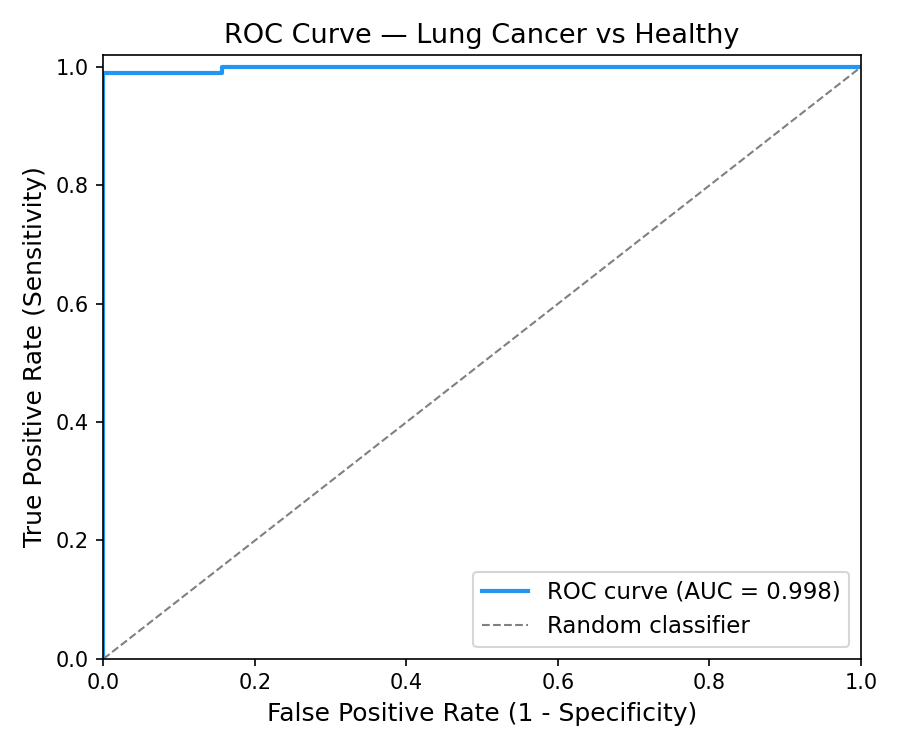
ROC curve of the model's predictions - understanding the relationship between false positives and true positives.
-
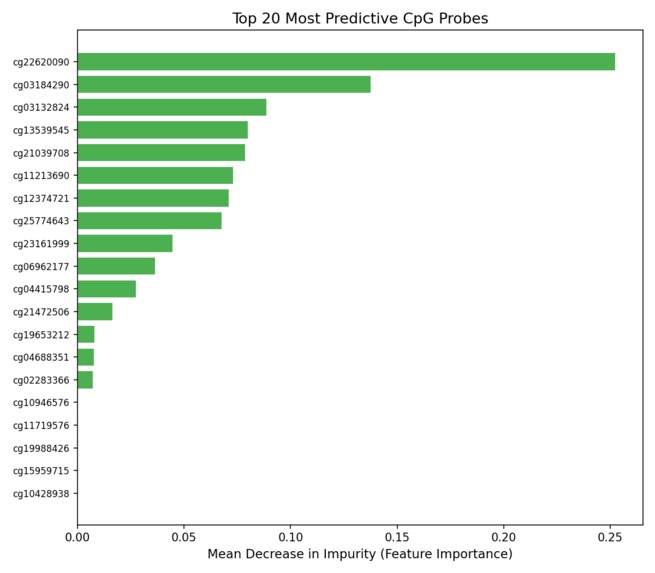
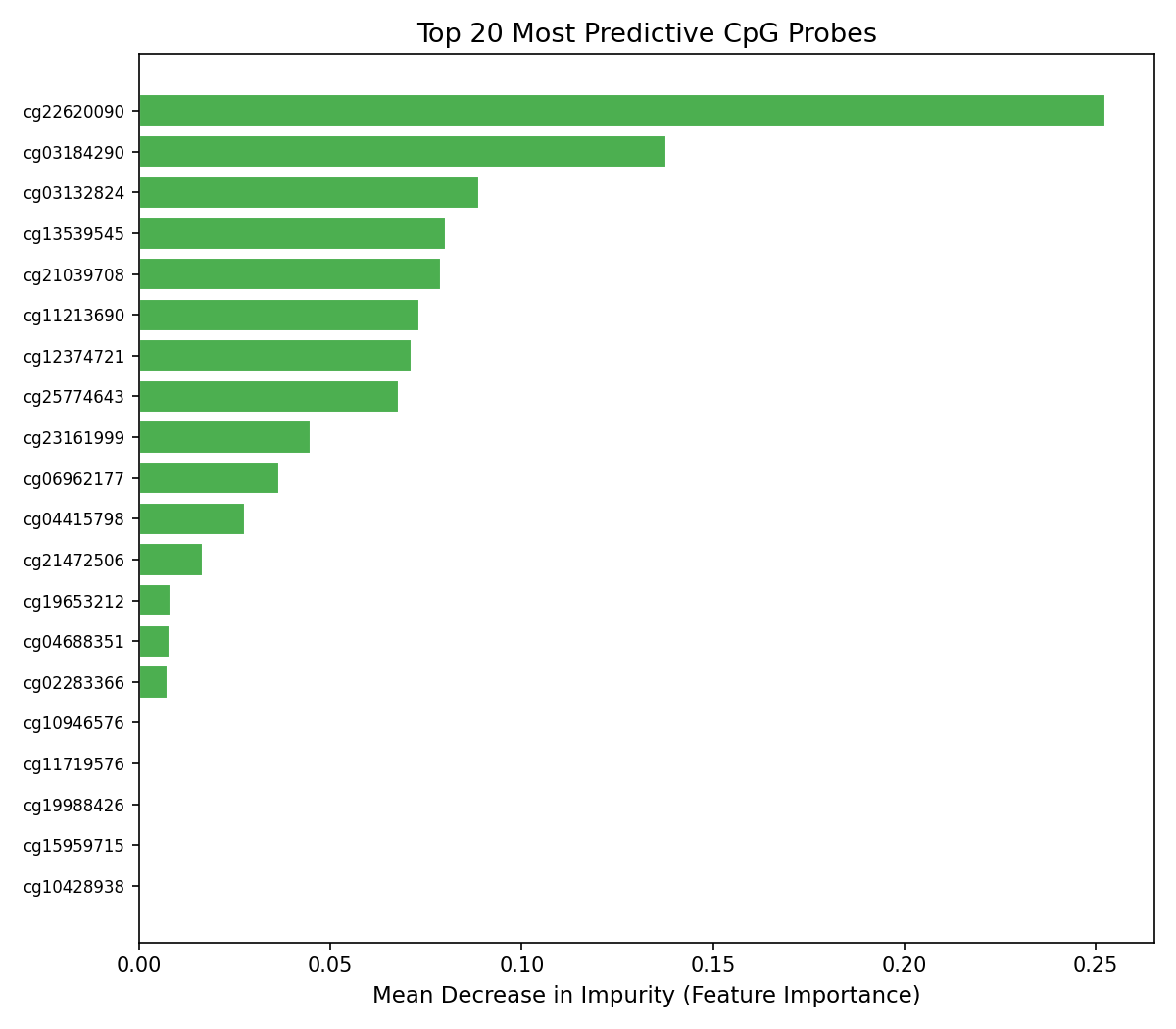
Feature Importance Graph - Which CpG sites are most used for prediction
-
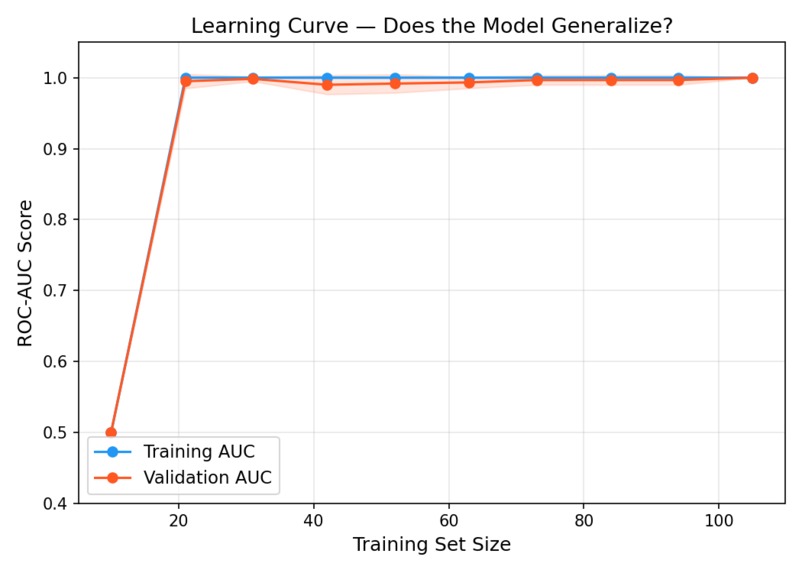
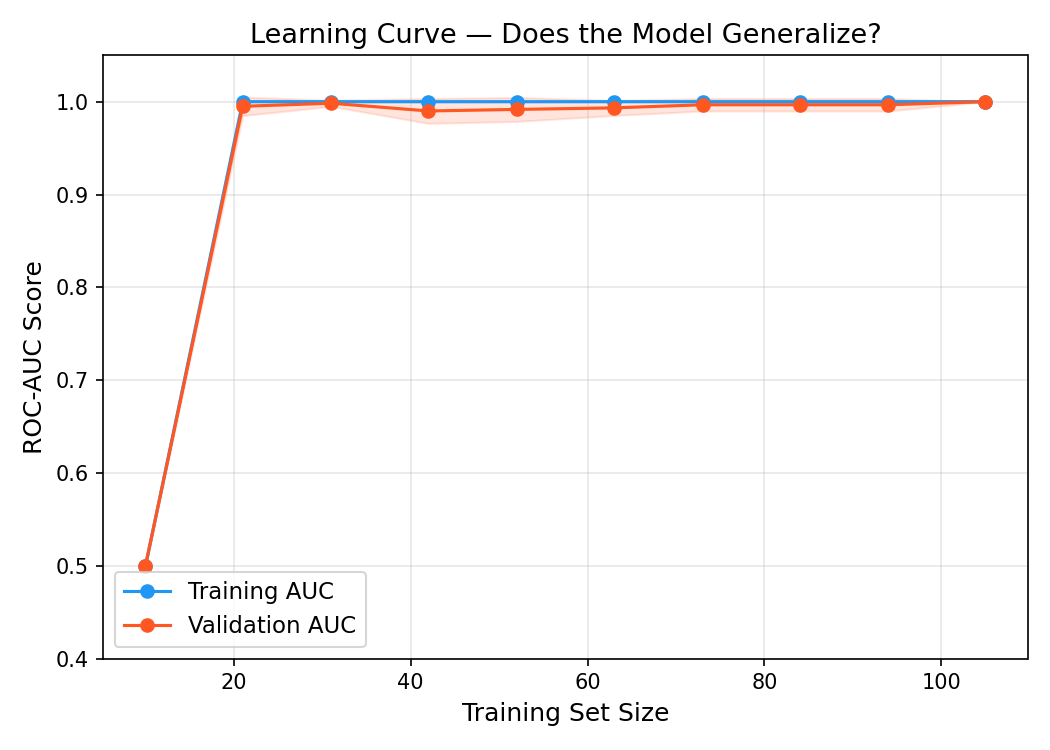
Learning curve to ensure model generalizes with new unseen data rather than memorize.
-
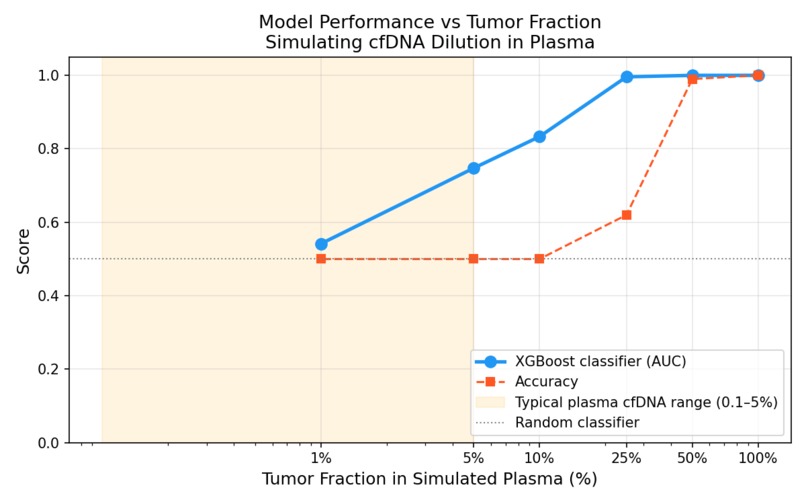
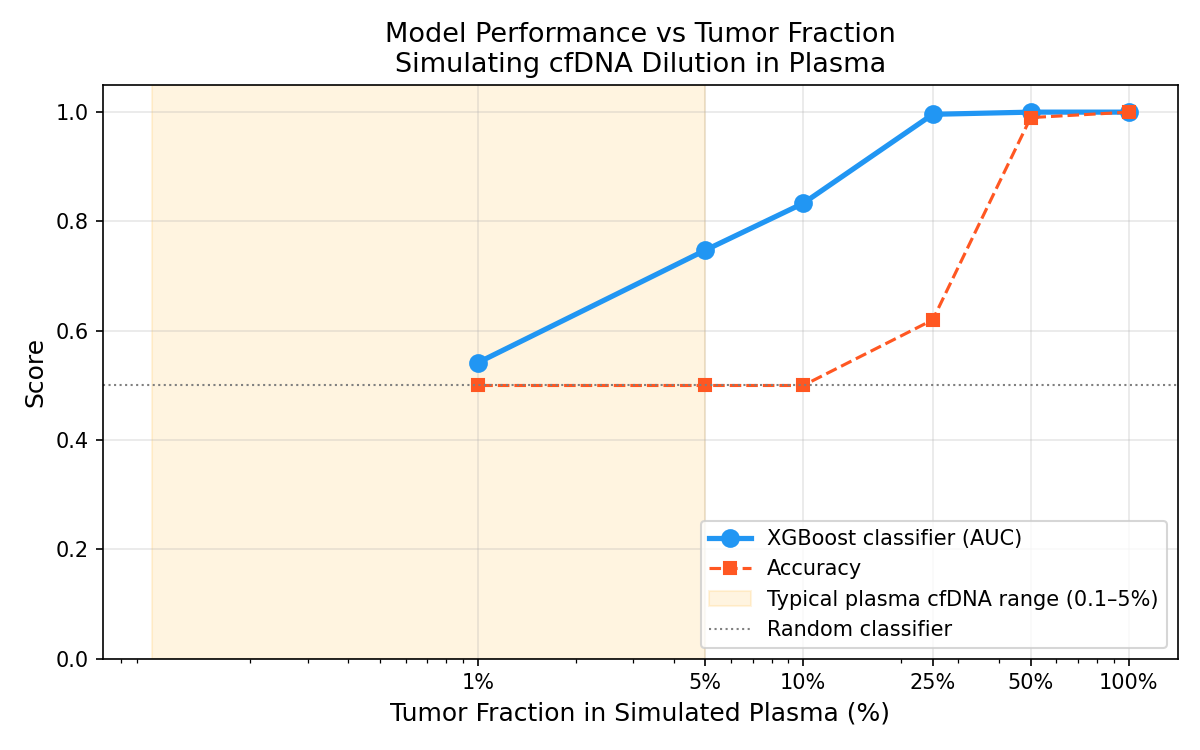
Tumor Fraction Graph - Understanding how the ROC changes with the percentage of healthy vs tumor sample in the dataset
Inspiration
21,000 Canadians die of lung cancer every year. The majority are diagnosed at Stage III or IV, when 5-year survival drops below 10%. At Stage I, it's above 80%. The difference isn't the disease, it's when you find it. Current screening tools like low-dose CT are expensive, expose patients to radiation, and produce high false-positive rates with extremely long wait times (average of 8.8 weeks). The field has been moving toward liquid biopsy: a simple blood draw that detects cancer-derived DNA circulating in the bloodstream. Takes upwards to 2 weeks to interpret. Companies like GRAIL have demonstrated it's possible. The question is whether I can build open, reproducible pipelines that researchers and clinicians can actually study and extend. That's what earlySignal is; an end-to-end proof-of-concept pipeline for lung cancer detection from cell-free DNA methylation profiles, built transparently so the limitations are as visible as the results. This project was strongly influenced by Dr. Fei Geng's work in liquid biopsy for early cancer detection at McMaster.
What it does
earlySignal takes DNA methylation array data, 450,000 CpG site measurements per patient sample, and classifies samples as lung adenocarcinoma (LUAD) or normal tissue using a gradient boosting model. The pipeline has three components:
- Classification: An XGBoost model trained on TCGA-LUAD data achieves 0.924 AUC under 5-fold cross-validation and 1.0 AUC on a held-out test set, demonstrating that tissue methylation signatures are highly discriminative.
- Interpretability: Feature importance outputs identify which CpG sites drive predictions, connecting model decisions back to known cancer biology.
- Tumour Fraction Simulation: The clinically honest part. I computationally simulated liquid biopsy conditions by mixing tumour and normal methylation profiles at decreasing tumour fractions (10% down to 0.1%). At 1% tumor fraction, the clinical reality of early-stage plasma cfDNA, model AUC degrades to 0.54. Essentially random. This quantifies exactly where tissue-trained models fail in the liquid biopsy context, and defines the problem that needs to be solved next. A Streamlit web app lets anyone interact with the model and explore the tumour fraction degradation curve interactively.
How I built it
- Data: TCGA-LUAD Illumina 450K methylation array data (132 samples, ~413,000 CpG probes), streamed from UCSC Xena using a custom downloader.
- Preprocessing: Probes with >20% missing values filtered out. Remaining missing values imputed with column means. Result: a clean 132 × 413,000 beta value matrix.
- Feature Selection: Custom TopVarianceSelector (scikit-learn compatible transformer) computes per-probe variance across training samples and retains the top 5,000 highest-variance probes. High variance = the probe behaves differently between tumor and normal; that's the signal.
- Model: XGBoost classifier wrapped in a scikit-learn Pipeline with the variance selector. Hyperparameters tuned for the dataset size to avoid overfitting on 132 samples.
- Evaluation: 5-fold stratified cross-validation, ROC/AUC curves, confusion matrix, learning curves, and feature importance plots, all in a dedicated evaluation module.
- Tumor Fraction Simulation: In silico mixing at 9 tumour fraction levels from 0.1% to 50%. AUC computed at each level to produce the degradation curve.
- Stack: Python 3.11, XGBoost, scikit-learn, pandas, numpy, matplotlib, Streamlit. Conda environment for reproducibility.
Challenges I ran into
The data size problem. 413,000 features and 132 samples is a high-dimensional, low-sample-size regime where models overfit trivially. Variance-based feature selection was necessary before any model could generalize and getting the selector to fit only on training data (not leak information from the test set) required careful pipeline construction. The honest result problem. The tissue classifier works almost perfectly. That's actually suspicious; it should be. Tissue biopsies are an easy version of the problem. Building the tumor fraction simulation was necessary to get to the real, hard number: AUC 0.54 at 1% tumor fraction. Presenting a result that shows your model failing is uncomfortable. It's also the most scientifically credible thing in this project. Feature interpretability. XGBoost gives you feature importances, but raw probe IDs (cg00000029, etc.) aren't meaningful on their own. Connecting the top probes back to gene annotations and known cancer loci required cross-referencing the 450K manifest, a step that took longer than expected.
Accomplishments that I'm proud of
By far, the tumor fraction curve. It would have been easy to stop at "AUC 1.0 on holdout, project done." Instead, the simulation shows exactly where and how a tissue-trained classifier breaks when applied to liquid biopsy conditions. That's a real scientific contribution, even at hackathon scale. The pipeline is also genuinely reproducible. Clone the repo, run three commands, get the same results. In a field where reproducibility is a known crisis, that matters.
What I learned
That the gap between a good classifier and a clinically deployable tool is enormous and the gap is precisely quantifiable. The 0.542 AUC at 1% tumor fraction isn't a disappointing result. It's a research finding: it tells you that signal dilution in plasma cfDNA is the binding constraint, and that solving it likely requires either training directly on plasma data, deconvolution models that estimate tumor fraction first, or feature selection specifically optimized for low-fraction detection. Also: building a proper ML pipeline from scratch, custom sklearn transformers, avoiding data leakage, and cross-validation done right is harder than I thought.
What's next for earlySignal - Liquid Biopsy Cancer Detection
The 30-day build period has a specific goal: external validation on real plasma cfDNA data. TCGA tissue biopsies are a starting point, not an endpoint. The next step is applying this pipeline to actual liquid biopsy datasets to see whether the model generalizes and what modifications are needed. GEO has several plasma cfDNA methylation cohorts available for this. Longer term: the pipeline is architected to be feature-agnostic. The variance selector and XGBoost classifier can accept any genomic feature matrix; k-mer frequencies, histone modification profiles, ATAC-seq accessibility scores. The goal is a config-driven framework where changing the feature type doesn't require rewriting the model or evaluation code.
Built With
- conda
- github
- illumina450k
- matplotlib
- numpy
- pandas
- python
- scikit-learn
- seaborn
- streamlit
- tcga-luad
- ucscxena
- xgboost


Log in or sign up for Devpost to join the conversation.