-
-

-
-
-
-
-
-
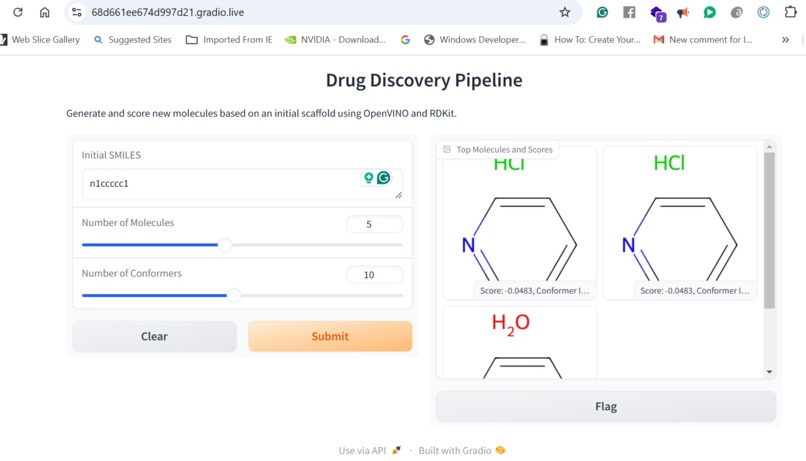
Creation of new Molecules using GenAI
-

Using Intel AMX usage in code
-
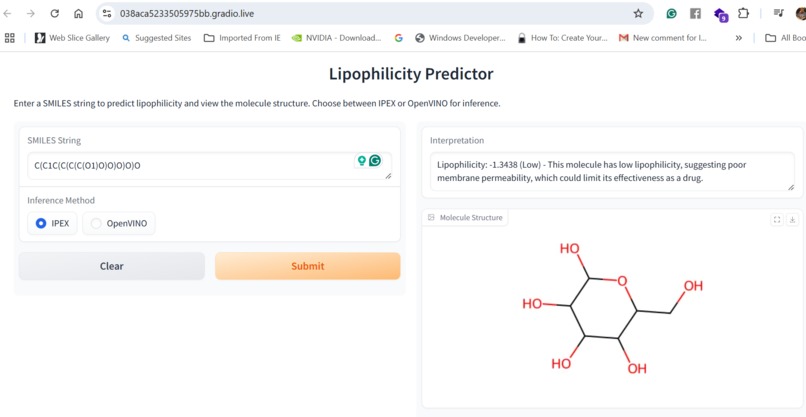
Inference using "IPEX" or "OpenVINO"
-

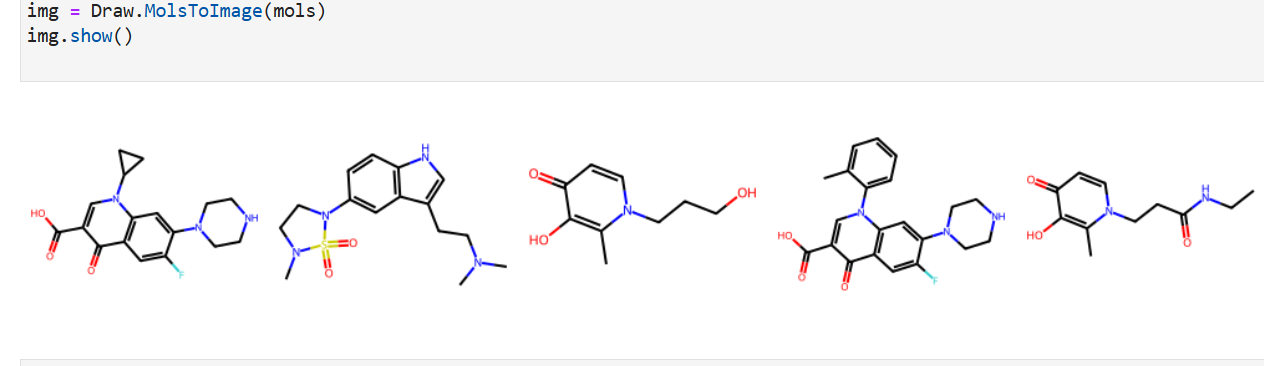

Inspiration
Drug discovery is a complex and costly process that can take years and billions of dollars to bring a new drug to market. With advances in AI, machine learning, and accelerated computing, we saw an opportunity to streamline the early stages of drug discovery significantly. We were inspired by the potential to integrate various AI-driven techniques—like predicting binding affinity, lipophilicity, and synthetic feasibility—to build a comprehensive platform that enhances decision-making in drug development. The availability of powerful, optimized frameworks like OpenVINO, Qdrant for vector-based storage, and Gradio for rapid deployment motivated us to build an application that leverages these tools for real-time predictions and decision support, accelerating research in a field that directly impacts human health.
What it does
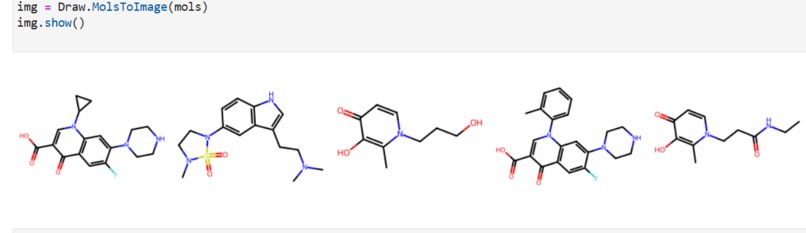
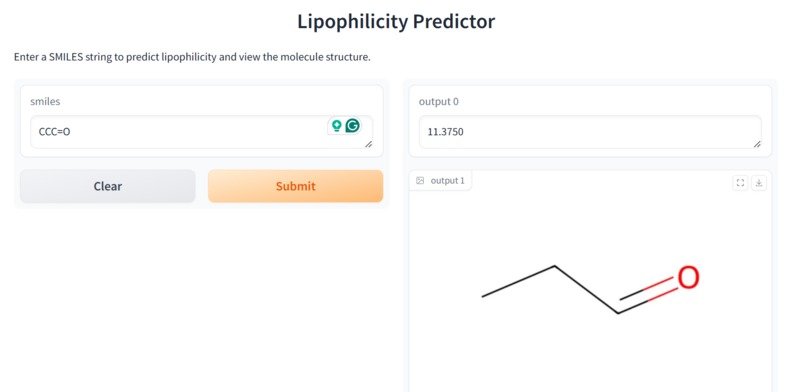
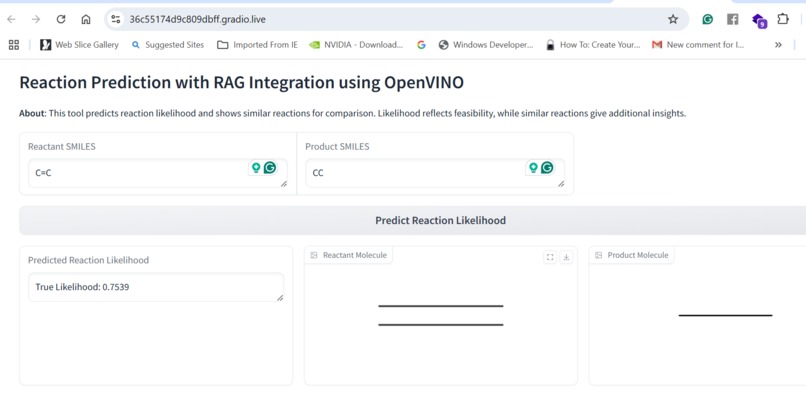
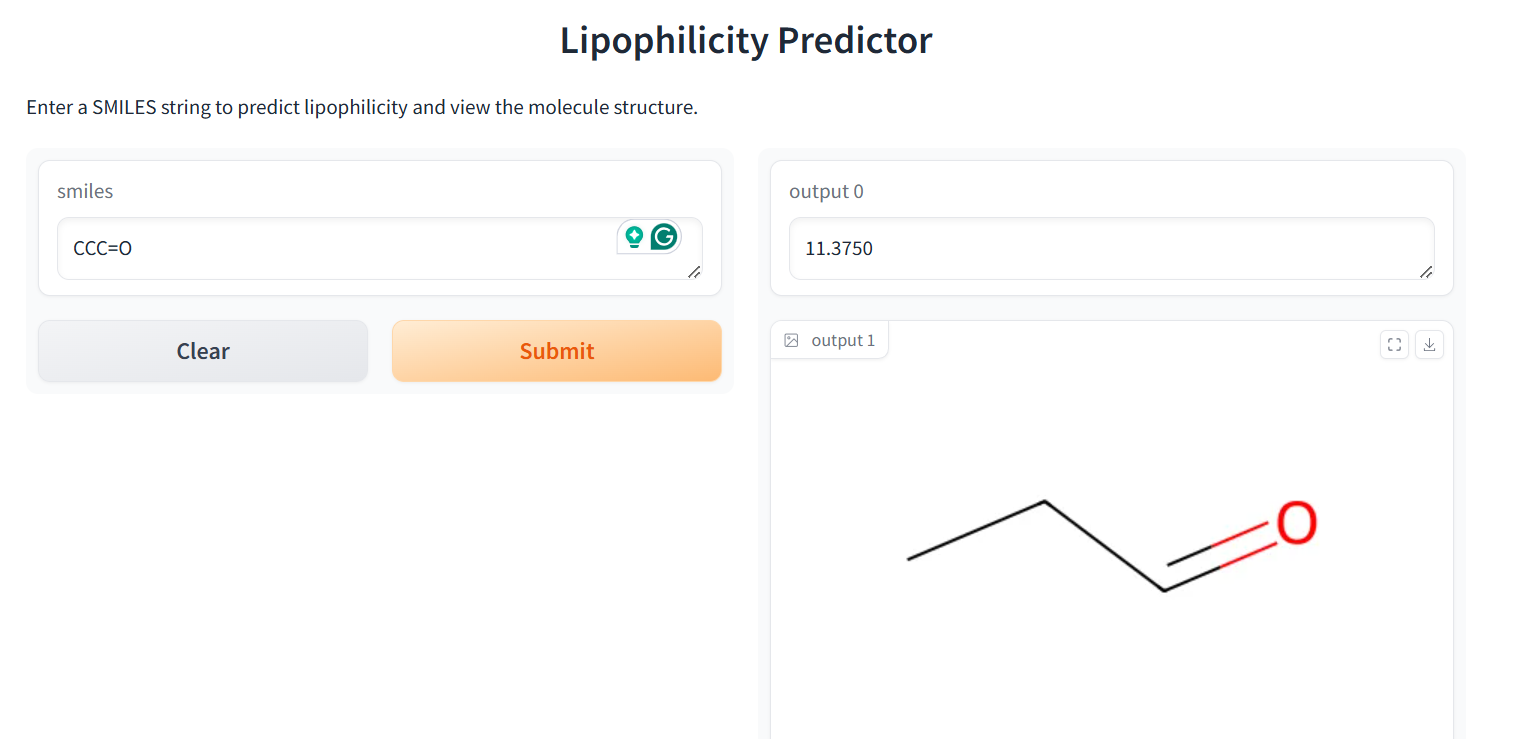
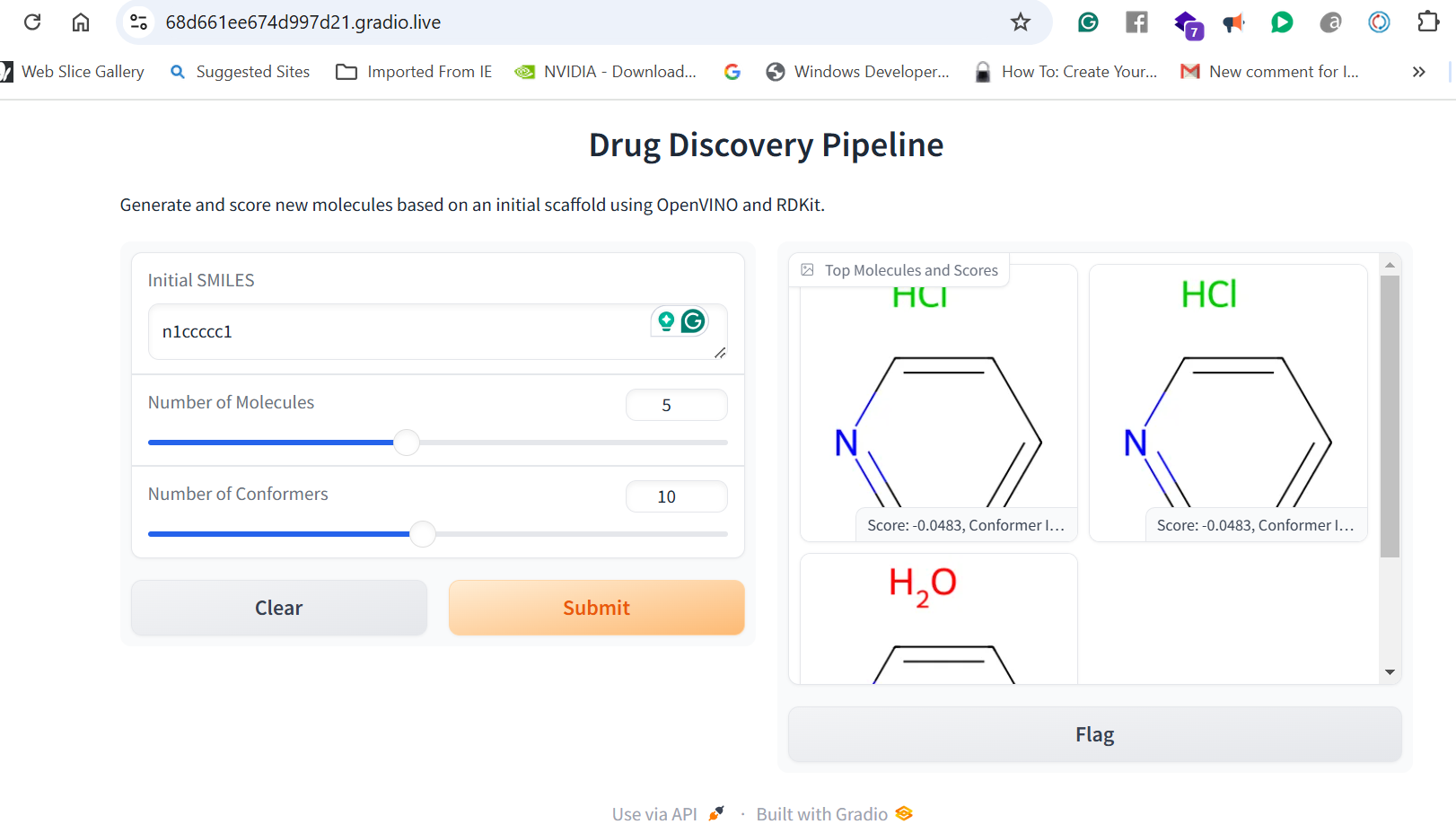
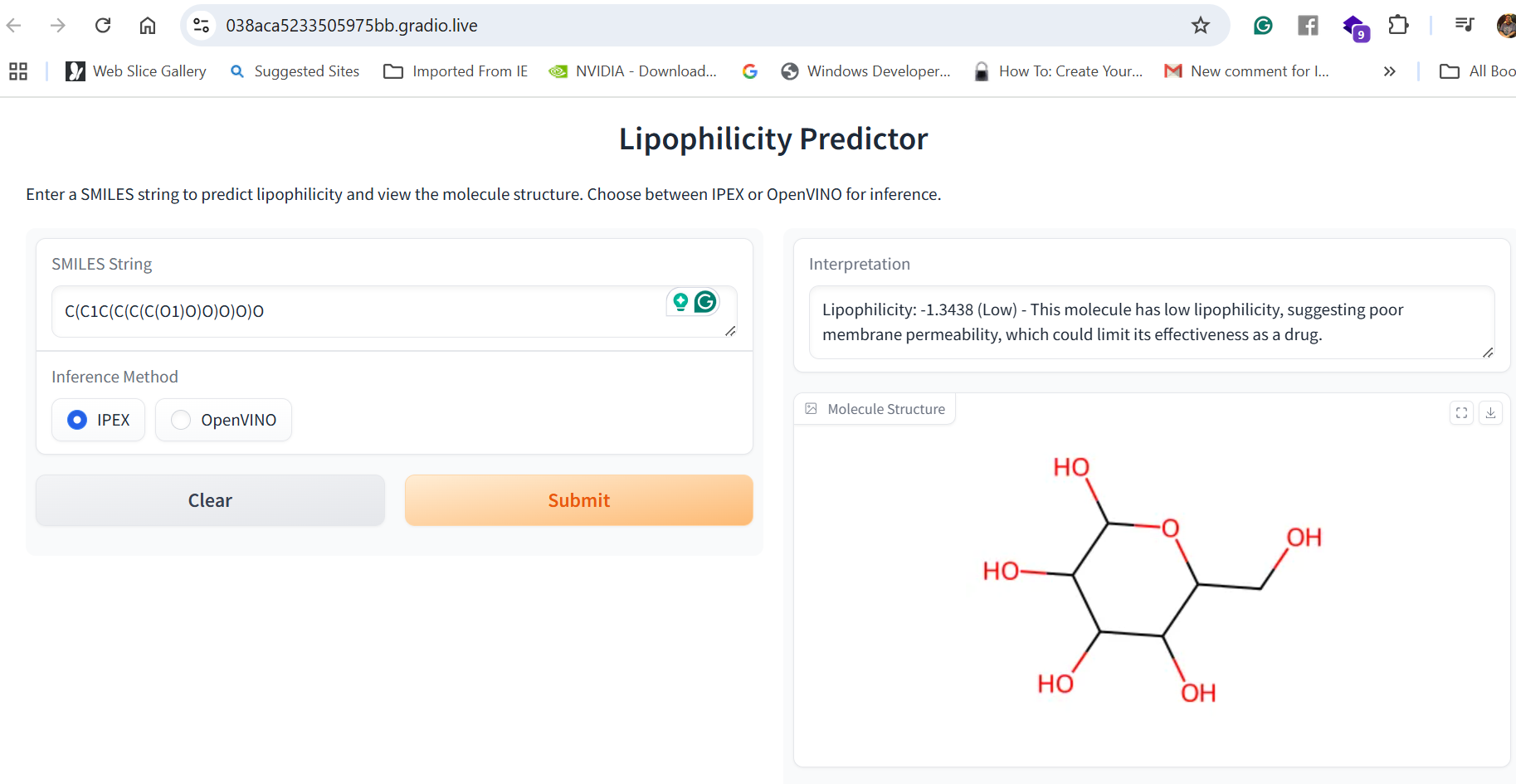
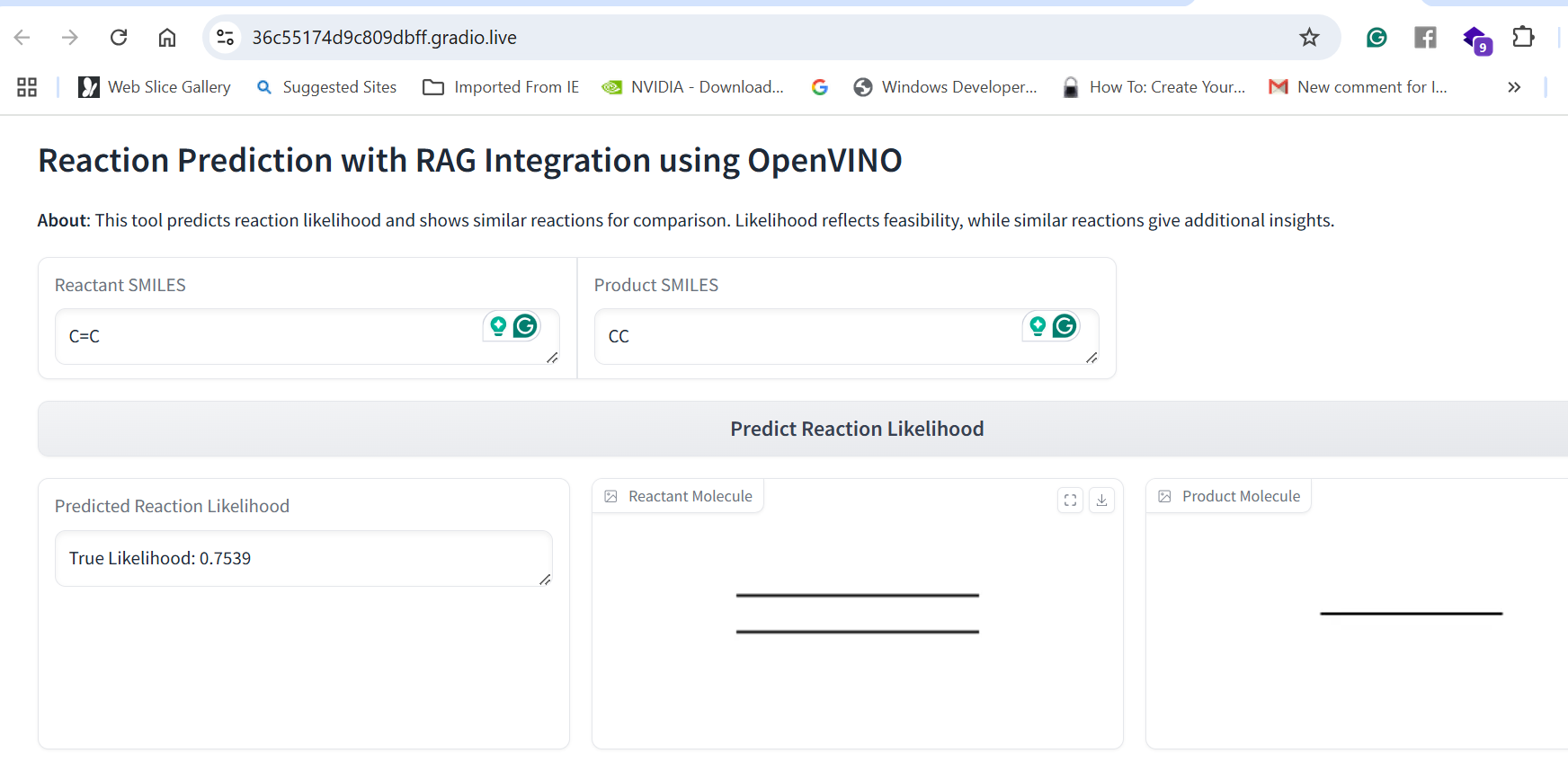
Our application combines multiple AI-driven steps in the drug discovery process, covering essential predictions and synthesis planning. It helps researchers in: Lipophilicity Prediction: Determining the hydrophobic or hydrophilic properties of a molecule, which influences its pharmacokinetics, such as absorption and distribution. Binding Affinity Prediction: Predicting the strength of interaction between a drug candidate and its target protein, providing insight into potential efficacy. Conformer Generation and Scoring: Generating 3D orientations (conformers) of molecules and scoring them for stability and binding potential, ensuring that the most viable molecular structures are prioritized. Reaction Prediction and Visualization: Predicting potential chemical reactions and visualizing molecular structures, which aids in planning feasible synthesis pathways. RAG-Enhanced Reaction Prediction: Utilizing retrieval-augmented generation (RAG) to integrate historical reaction data with model-based predictions, improving the accuracy and relevance of synthetic pathway suggestions. The application is an AI-powered assistant, allowing researchers to evaluate drug candidates, generate synthetic pathways, and optimize drug properties, accelerating the entire drug discovery pipeline.
How we built it
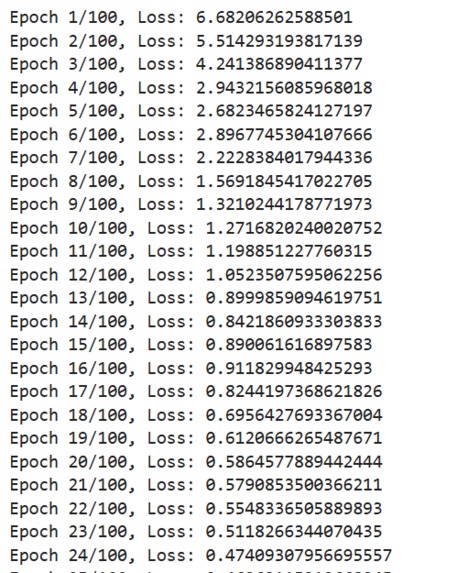
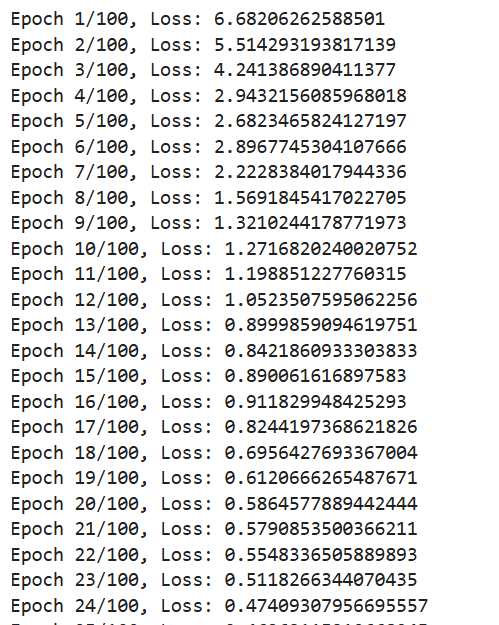
Data Collection and Preprocessing: We gathered molecular datasets containing SMILES representations for drug molecules, lipophilicity values, binding affinity scores, and known reaction data for training and validation. Using RDKit, we generated molecular fingerprints and conformations, allowing the model to use numerical vectors that represent structural and functional characteristics. Model Training and Optimization: Models were trained to predict lipophilicity, binding affinity, and reaction likelihood based on historical data. Each model was then optimized with OpenVINO for efficient CPU-based inference, making it possible to achieve fast, real-time predictions. Reaction prediction was enhanced by integrating a RAG pipeline with Qdrant as the vector database. This setup allows the application to retrieve similar reactions, offering historical context and improving prediction quality. System Integration: OpenVINO: Optimized models for inference to ensure high-speed, CPU-efficient predictions suitable for real-time applications. Qdrant: Deployed as an in-memory vector database for storing and retrieving reaction fingerprints, enabling efficient similarity searches. Gradio: Used for creating an interactive web-based interface that allows users to input molecular data, run predictions, and view results directly, facilitating ease of use and rapid testing. Deployment: We containerized the entire application and deployed it on OpenShift, enabling scalability and parallel processing for high-throughput drug discovery applications.
Challenges we ran into
Data Quality and Availability: Ensuring we had high-quality, diverse molecular data to train models effectively was challenging. Some molecular datasets lacked labels for lipophilicity or binding affinity, necessitating further curation. Model Optimization: Adapting and optimizing complex models for OpenVINO to ensure high-speed inference without sacrificing accuracy required significant tuning and validation. Integrating RAG: Designing an efficient RAG pipeline that could retrieve relevant historical reactions from Qdrant and feed them into the prediction pipeline without introducing latency was a technical hurdle. Interface Design: Creating an intuitive Gradio interface that supports complex workflows, yet remains user-friendly, involved extensive iterations and testing to ensure the UI met both functional and accessibility requirements. Deployment Scaling: Deploying on OpenShift and configuring resources to handle large-scale requests was challenging, particularly in optimizing container resources for parallel predictions.
Accomplishments that we're proud of
Real-Time Prediction Capability: We successfully achieved real-time predictions for reaction likelihood, binding affinity, and other drug discovery properties, making the application fast and practical for researchers. RAG Integration: We’re proud of our RAG-enhanced pipeline, which leverages historical reaction data to improve prediction accuracy, providing researchers with contextually relevant synthesis pathways. Comprehensive Workflow: By integrating multiple drug discovery stages into a single application, we created a versatile tool that can address multiple aspects of early drug discovery, saving time and resources. Scalable Deployment: Deploying the application on OpenShift with container orchestration means it can handle high-throughput workflows, making it suitable for both small-scale testing and large-scale drug screening. User-Friendly Interface: With Gradio, we developed an accessible interface that allows researchers to input data, view predictions, and retrieve relevant information effortlessly, lowering the barrier to using advanced AI in drug discovery.
What we learned
The Value of Data in AI-Driven Drug Discovery: Quality and diversity of data are fundamental for accurate predictions in drug discovery. We learned the importance of carefully curated data, as model performance heavily depends on the datasets used for training. Optimizing for Speed and Accuracy: OpenVINO was instrumental in helping us balance speed and accuracy. We gained hands-on experience in model optimization for CPU-based inference, learning how to achieve real-time predictions even on complex data. Importance of Context in Predictions: The RAG pipeline taught us that context from historical data can enhance model predictions, particularly in reaction prediction. This approach improves not only the prediction accuracy but also the interpretability and relevance of the results. Scalable Application Deployment: Deploying on OpenShift provided us with insights into scaling containerized applications and managing resources effectively for parallel processing in high-throughput environments. Creating Intuitive AI Tools: Building a user-friendly interface for complex AI workflows reinforced the importance of designing accessible tools for end-users, especially in domains like drug discovery where specialized knowledge is required.
What's next for Drug Discovery using AMX and OpenVINO
Expand Dataset and Model Training: We plan to incorporate more datasets covering additional properties like toxicity, permeability, and metabolic stability, further enhancing the platform’s predictive power. AMX Integration: By integrating AMX (Advanced Matrix Extensions) with our current OpenVINO setup, we aim to take advantage of Intel’s latest hardware acceleration capabilities, further speeding up computation and handling more complex models in real-time. Refining Conformer Generation and Screening: We will improve the conformer generation pipeline to explore more molecular orientations and enhance the scoring algorithm, helping to better capture 3D structures that are important for drug efficacy. Automated Feedback Loops for RAG: Enhancing the RAG pipeline to allow dynamic updates from newly generated reaction data or published literature could make predictions even more accurate over time by continually learning from the latest data. Improved Deployment and Scaling on OpenShift: We’ll explore ways to further optimize our deployment on OpenShift, leveraging Kubernetes-native tools to manage resources more efficiently, particularly for high-demand applications like large-scale compound screening. Extended User Interface Features: We plan to add more interactive features to the Gradio interface, such as detailed visualization of molecular interactions and user-adjustable parameters for fine-tuning predictions, making the tool even more adaptable to researchers' needs. Collaboration with Pharma Industry: We aim to collaborate with pharmaceutical researchers and institutions, gathering real-world feedback to refine the application and ensure it meets the needs of professionals in the drug discovery field.











Log in or sign up for Devpost to join the conversation.