-

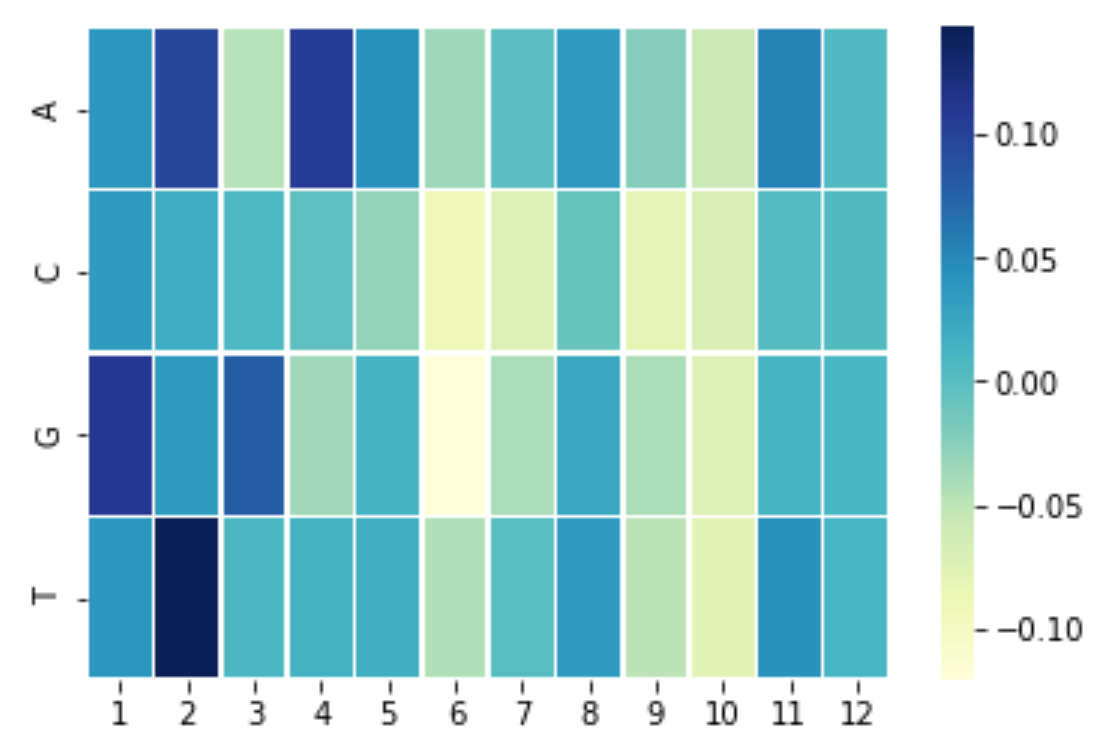
Attribution of the ContextNNN model
-

context example

ContextNNN (Context Nucleotide Neural Net)
Inspiration
DNA sequencing and genotyping have been widely adopted as a critical component of medical diagnostic, prognostic, and drug development. In some critical diseases, knowing the genetic variant would lead to better therapeutic plan or saving life. Although, genotyping has been widely adopted, detecting or determining the correct form of the variant are challenging especially variants around the repetitive regions and segmental duplication.
Since the context around the variant could affect the genotyping, measuring their impact on genotyping accuracy would enable us to develop a better variant caller algorithm. Neural network and deep learning provide us the framework to learn very complex structure without predefined features, so it is an attractive framework to assess the impact of genomic context on genotyping.
What the model does
We formulated our problem as followed. Given the base pair in the reference genome around the variant site (defined as context bases), can we predict if the variant caller will report the correct variant? In the final model, the context bases are the 12-mer to the right of the variant according to the reference genome. For the rest of the description, we will refer to this context bases as 12-mer context.
Dataset
We used variant calling from an individual with code HG002 using DeepVariant tool because DeepVariant tool has been showed to achieve the highest F1 score among popular variant callers (See https://blog.dnanexus.com/2017-12-05-evaluating-deepvariant-googles-machine-learning-variant-caller/). The variants from DeepVariant were then labeled with correct or incorrect call using the truth set from The Genome In A Bottle Consortium (https://www.nist.gov/programs-projects/genome-bottle) for HG002 individual. We removed the single nucleotide variants from the input set and focus only on the insert/deletion because single nucleotide variants are now detected with extremely high accuracy by most variant calling programs. Therefore, there is more room for improvement for insertion/deletion type of variant.
How I built it
We started our prototype using Keras and transitioned into TF2.0. We use functional API for the model rather than the sequential even though the current model does not include any branching because the functional API would allow us to expand our model into a more complex one later.
We designed the model with basic Dense network rather than the CNN network because we do not try to find a motif of particular size within our 12-mer context. We start with four layers of network with twelve nodes in each layer and try to reach the highest possible accuracy in the training set. Then we trimmed the model down to two layers while keeping the same level of accuracy. The class weight was used to handle the imbalance data.
Challenges I ran into
The major challenge for this study is a limited number of input. In a preliminary study, we compare the regression approach and classification approach. The regression approach would try to predict the ratio of correct variant calling for each unique 12-mer context, while the classification approach would give a correct/incorrect label to the each 12-mer context. The regression approach sounds more promising; however, this require us to collapse input with the same unique 12-mer context into one input which shrinking down the total input size. Also, it would be hard to partition data into training and test set with the same distribution.
Formulating the model into classification approach is also beneficial because we can calculate the lowest possible error or Bayes error which can be used as a guide to determine if the model performance on the training has reach the highest possible level of performance.
Since we can give only one label to each unique 12-mer context, the Bayes error can be defined as 1-sum(max(concordant_i, discordant_i))/total case; where _i denote the count of concordant/discordant for each unique 12-mer context. For this study, the Bayes error is 26%, so the highest possible accuracy is 74%.
Accomplishments that I'm proud of
After a few iteration of hypermeter adjustment, we are able to get the error in training, validation, and test set close to the Bayes error.
What I learned
Most of the effort in deep learning is indeed to defining the input/output, prepare the data, and set the environment for execution. Writing code for the actual deep learning part is much simpler.
Model inspection is also very important. In an earlier version of the model, our model stuck around moderate accuracy. After inspection, we found that the model overfit to the class with higher representation. Therefore, we include class weight to counteract the imbalance class.
What's next for ContextNNN
The main caveat for the current attribution analysis is that we analyzed the variant calling from ContextNNN. Most variant caller use explicit statistic model that take into account the quality/quantity of the mapped reads, while ContextNNN use the CNN model that capture mapped reads at the variant site and all the surrounding context. We believe that this lead to uninterpretable of attribution analysis. Therefore, one major goal for the next step of this project is to experiment on other variant calling tool.
The second issue to further investigate is the size of the context bases. In this study, we only use the context on the right of the variant calling site and only experiment on 12-mer. Different k-mer size would affect the binning of the data into a group and assign value for prediction, so this would directly affect the model. We would like to expand the analysis to include both left and right contexts and vary the size of the k-mer.
Third, we plan to use more TF2.0 component including TF.data rather than basic numpy or custom loss function with TR probability rather than using weight for imbalance class. The rational for the transition into TF.data is to promote the usage of sharable standard data structure in the bioinformatics community. For TR probability, it would make more sense to set loss function of the model to F1 score which is ready for interpretation of compare across the models.

Log in or sign up for Devpost to join the conversation.