-
-

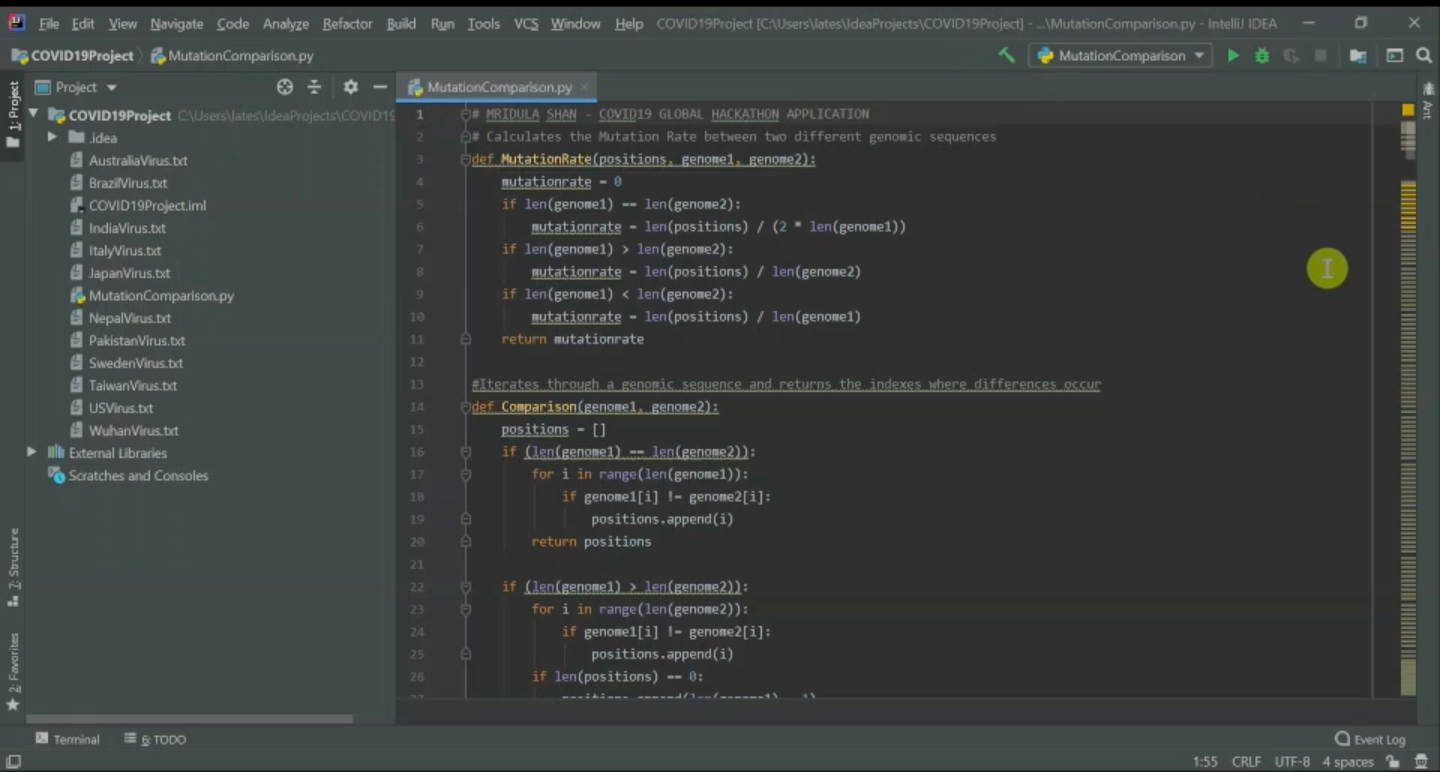
Screenshot of my IDE and Program
-

Stacked Bar Chart of the Mutation Rates
-

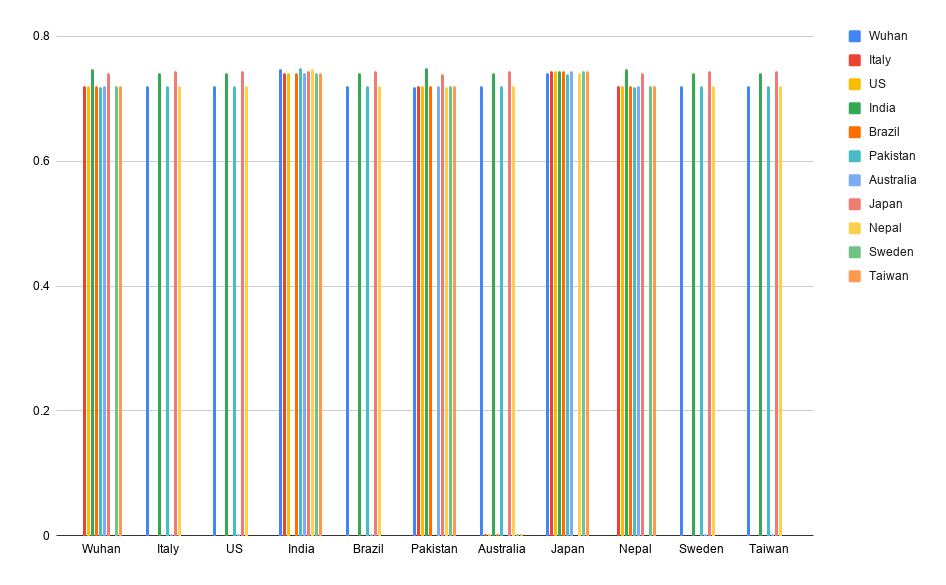
Data Collected from the Program comparing the Eleven countries
-

Column Chart for the Mutation Rate
-

Screenshot of the Output of the Program



Inspiration
I was inspired to create this program after learning more about how viruses spread throughout populations and species, and why the SARS-CoV-2 virus was unique in its need for the implementation of social distancing. After learning more about mutations, I learned that these changes are necessary for the evolution of viruses and their transformation to be viable in other organisms and to fight against varying immune systems. In essence, the variations of the virus help it survive in its various hosts. However, SARS-CoV-2 is unique in that although it mutated from bats, it mutates relatively slowly in humans and throughout different populations. Yet we still need social distancing in place in order to prevent the virus from being able to enter more hosts and adapt to the varying conditions of each environment.
What it does
The Mutation Rate Calculator is able to compare the varying genomic sequences of different strains of the virus from around the globe. It has two functions/methods: The comparison function iterates through both genomic sequences and compares each base to see if they are equal. If they are not, that position is added into an array that saves all the positions in the genomes where there are differences in the base pair. The MutationRate function calculates the mutation rate from the array generated before, by dividing the length of the arary which represents the total number of mutations by the length of the smallest genome or two times one of the genome's length if they are of the same length. The program then runs all various combinations of two genomic sequences from the eleven sequenced genomes available from the National Center for Biotechnology Information and outputs to the user the positions of where the base sequences were different along with the mutation rates calculated from such positions.
How I built it
I developed the program using the python programming language and intelliJ IDE, with the text files for the genomic sequences hardcoded into the program and formatted from the sequences provided by the National Center for Biotechnology Information.
Challenges I ran into
At first, it was difficult to develop the logic iterating through the entire sequence, because I had to take into account conditions where one genomic sequence length was greater than the other, since if such mutations/variations occur, it is probable that there would be greater variations in the length of the entire genomic sequence as well. Likewise, if the larger genomic sequence would be determining how far the program was to iterate then errors would occur because the smallest genomic sequence doesn't even have those characters to compare it to. However, I was able to understand such logic by taking into account that any nucleotide base over the length of the smallest genomic sequence is automatically a mutation and so is every nucleotide after it. Therefore, by taking into account that the largest genome would have more mutations in it in comparison with the original genomic sequence, my final code just had to incorporate those conditionals to test for the larger length and iterate only up until the total length of the smallest genome and adding all the other additional bases from the larger genome.
Accomplishments that I'm proud of
I am a high school student, and I am proud that I am able to contribute to this worldwide effort of supporting communites and understanding more about SARS-CoV-2, while researching how to stop its effects on our communities.
What I learned
I learned that mutations of the coronavirus over various strains are not the only factor determining how it is able to sustain in various environments and spread amongst different populations. After researching more about mutations, I learned that the dN/dS ratios, that is the synonymous versus nonsynonymous mutations, of the virus also play a role in how these mutations aid in the evolution of the virus over time. This got me interested in a greater expansion of the project in order to understand how these mutations actually affect the virus's ability to survive and reproduce in various environments and why certain mutations are more likely to occur in one strain of the population in comparison to another.
What's next for COVID-19 Mutation Rate Application
For my application, I want to create another function that is able to calculate the dN/dS ratio of the virus in order to see how the virus is evolving and in what way, as in wether or not the evolution of the virus is providing it with an advantage to survive longer in harsher environments or if it is becoming weaker and slowly dying out. What's more, I want to add in a visual representation to the project, such as generating its own graphs through the program, rather than relying on excel to create the graphs from the mutation rates calculated by the program. Likewise, I would like to add a graphical user interface for users to be able to input their own variations of the virus or another two genomic sequences that they would like to compare rather than relying on hardcoding the textfiles into the program and having the run command in the IDE showcase the variations to the user.



Log in or sign up for Devpost to join the conversation.