-
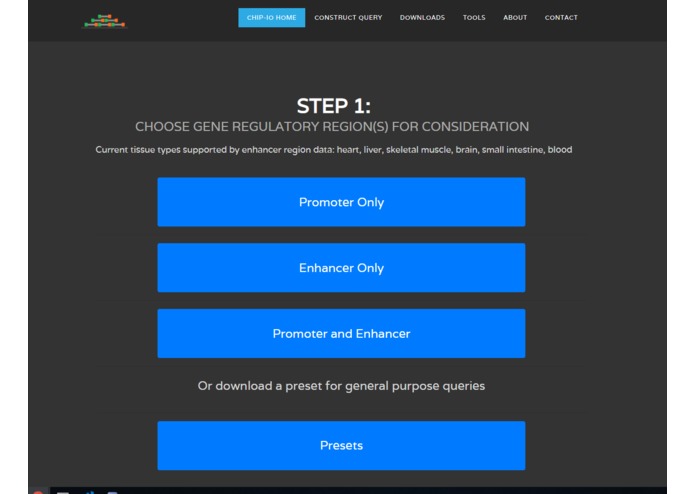
Analysis Query Form
-
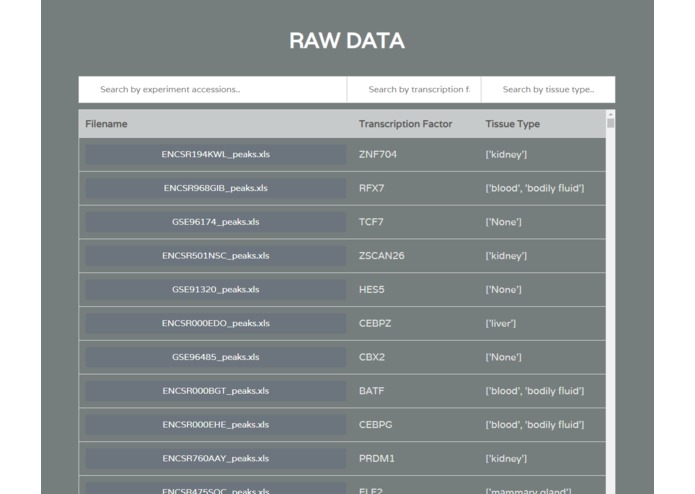
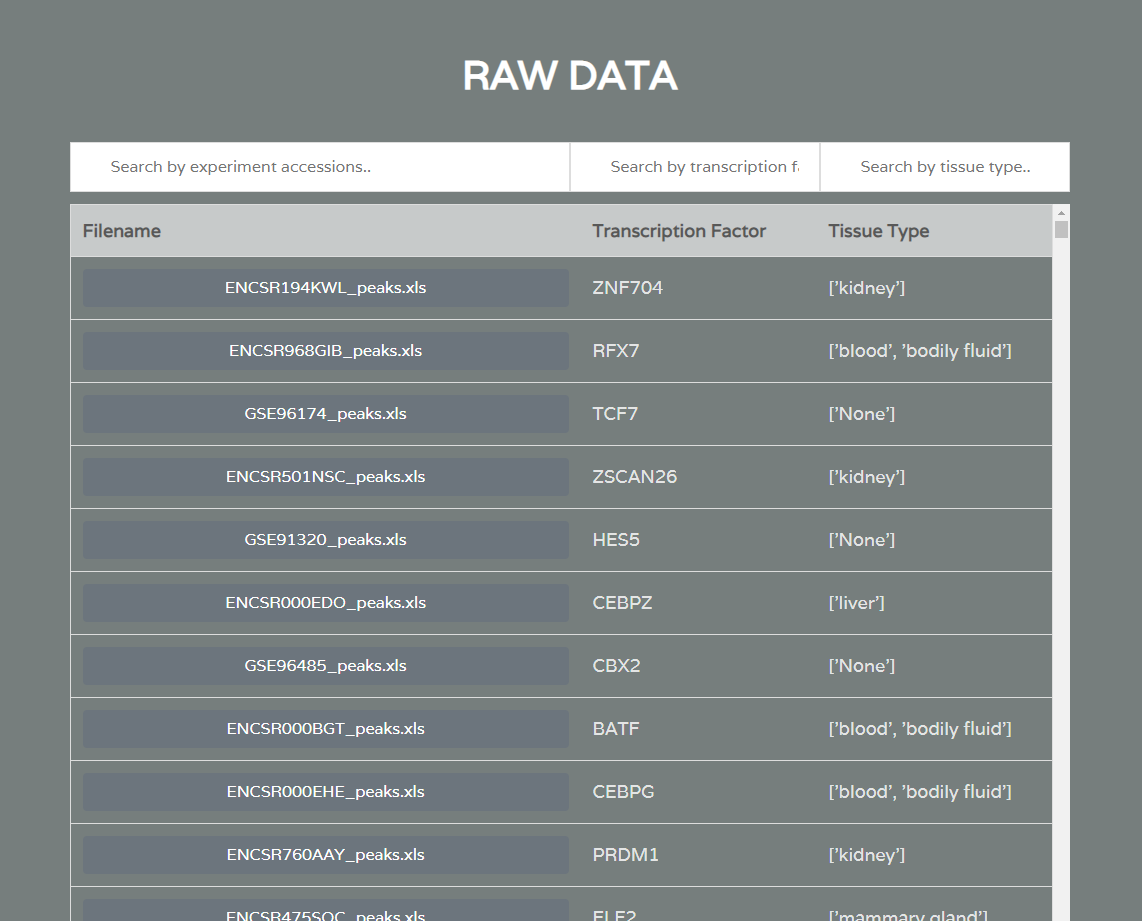
Raw Data Download
-
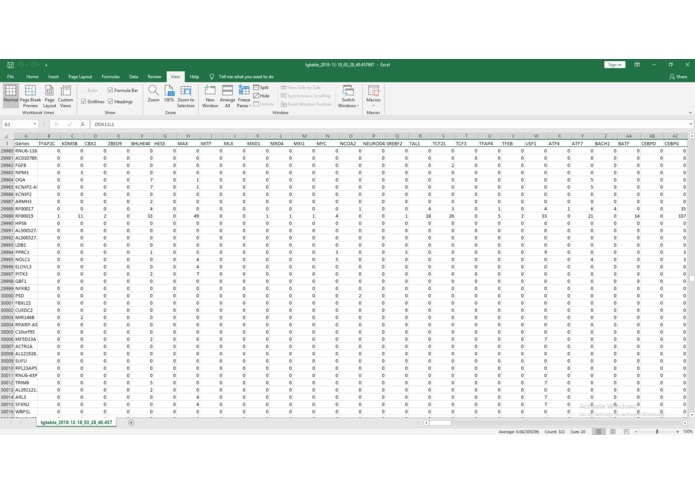
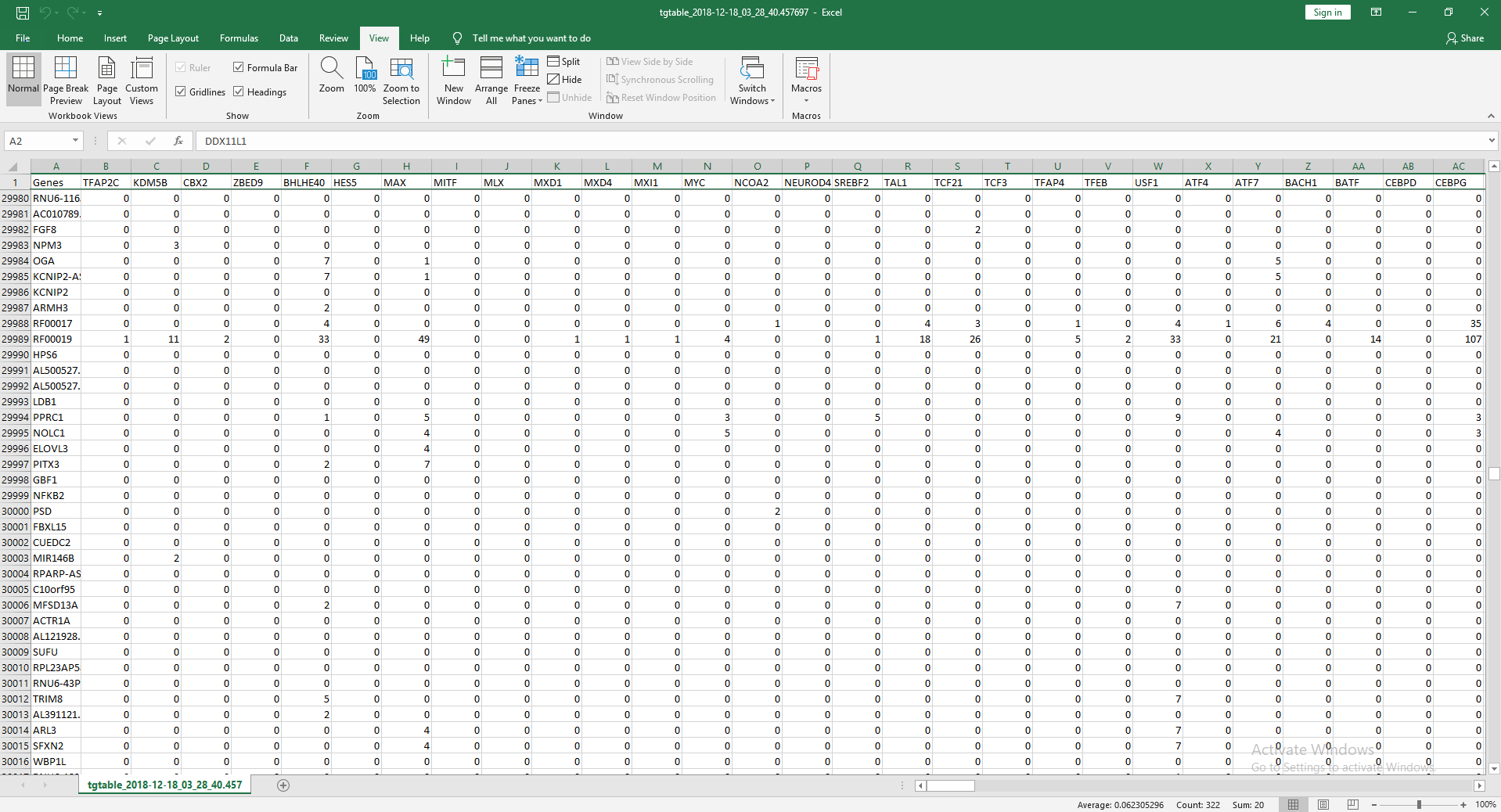
Example Output Table
Inspiration
A common need among bioinformations and computational biologists is accurate mapping of which transcription factor proteins regulate which genes. Unfortunately, the answer to this question depends on numerous factors including cell/tissue type, developmental phase, and even environmental conditions. If I had to guess, scripts for answering this question have been independently developed and used across research groups and individuals. Many are likely out-of-date. ChIP-IO attempts to consolidate this task, helping to push the field forward. As an added detail, most bioinformatics tools are notoriously lackluster in terms of user-friendliness and general appeal. In my opinion this is a weakness in the field, likely due to the lack of funding for full-time web developers in research labs. ChIP-IO sets a precedent by more closely following prevailing web design standards with an emphasis on user-freindliness.
What it does
A separate data collection and standardization step was done to build the app database. This involved the automated querying of ChIP-Seq data from GEO and ENCODE, two of the largest repositories of biological data. Metadata was extracted for each study and bioinformatics processing was done to generate standardized ChIP-Seq peaks.
Due to the limited numbers of TFs and tissue types which have been experimentally profiled thus far, ChIP-IO will also include in silico predicted TF binding locations.
ChIP-IO hosts this data for direct download, but more importantly allows for custom gene regulatory analysis. Users can submit query forms containing parameters such as tissue type, p-value cutoffs for ChIP-Seq peaks, promoter region definitions, etc. Once submitted, the back-end will apply constraints, assemble custom gene regions, and perform the necessary operations mapping ChIP-Seq peaks to known, gene-specific regulatory regions. These results are than returned as downloadable table files mapping TFs to genes.
How I built it
Data collection was done through python pipelines designed to be run on linux clusters in parallel. Intermediate operations were done using common bioinformatics command line tools where appropriate. Metadata processing was done using the ENCODE API for studies sourced from ENCODE. Due to GEO's poor data standardization, metadata was first mined using the Entrez API followed by semi-manual parsing in Python.
The web-app itself was built using the Python-flask framework. The front-end was built on top using just HTML, CSS, Javascript, and Bootstrap+jquery. The back-end is in python with a healthy dose of command-line utilities and bioinformatics tools for doing the information processing. No formal database was used as many of the utilities assume files as input and the benefits afforded by a database are somewhat marginal here. The app was hosted on an Ubuntu AWS EC2 instance with a Gunicorn application server and NGINX reverse-proxy server.
Challenges I ran into
Automated data collection is not nearly as simple as it sounds, especially when attempting to scale on clusters. There are also a huge number of metadata inconsistencies, errors, and missing data which have to be accounted for in the automation process. GEO data is especially hard to reconcile.
This is my first time constructing a web application truly from scratch, especially one of this magnitude. The biggest mistake was not developing using a python virtual environment. This is a great way to keep track of only what the application needs to run. There were also many smaller issues with figuring out how to properly deploy the application (hint:don't use heroku for anything heavy duty), and other aspects of cloud computing and networking which are unfamiliar to me.
Accomplishments that I'm proud of
Everything.
What I learned
Quite a bit.
What's next for ChIP-IO
The biggest next step is incorporating in silico TF predictions to expand the database substantially. I also need to write up a publication to get the word out.

Log in or sign up for Devpost to join the conversation.