Chemical_Structure_Reconstruction
Project submission for Pharmahacks (2019)
The Task
We developed an algorithm that can identify the least amount of Mordred descriptors needed to reverse engineer chemical structures.
Our Approach
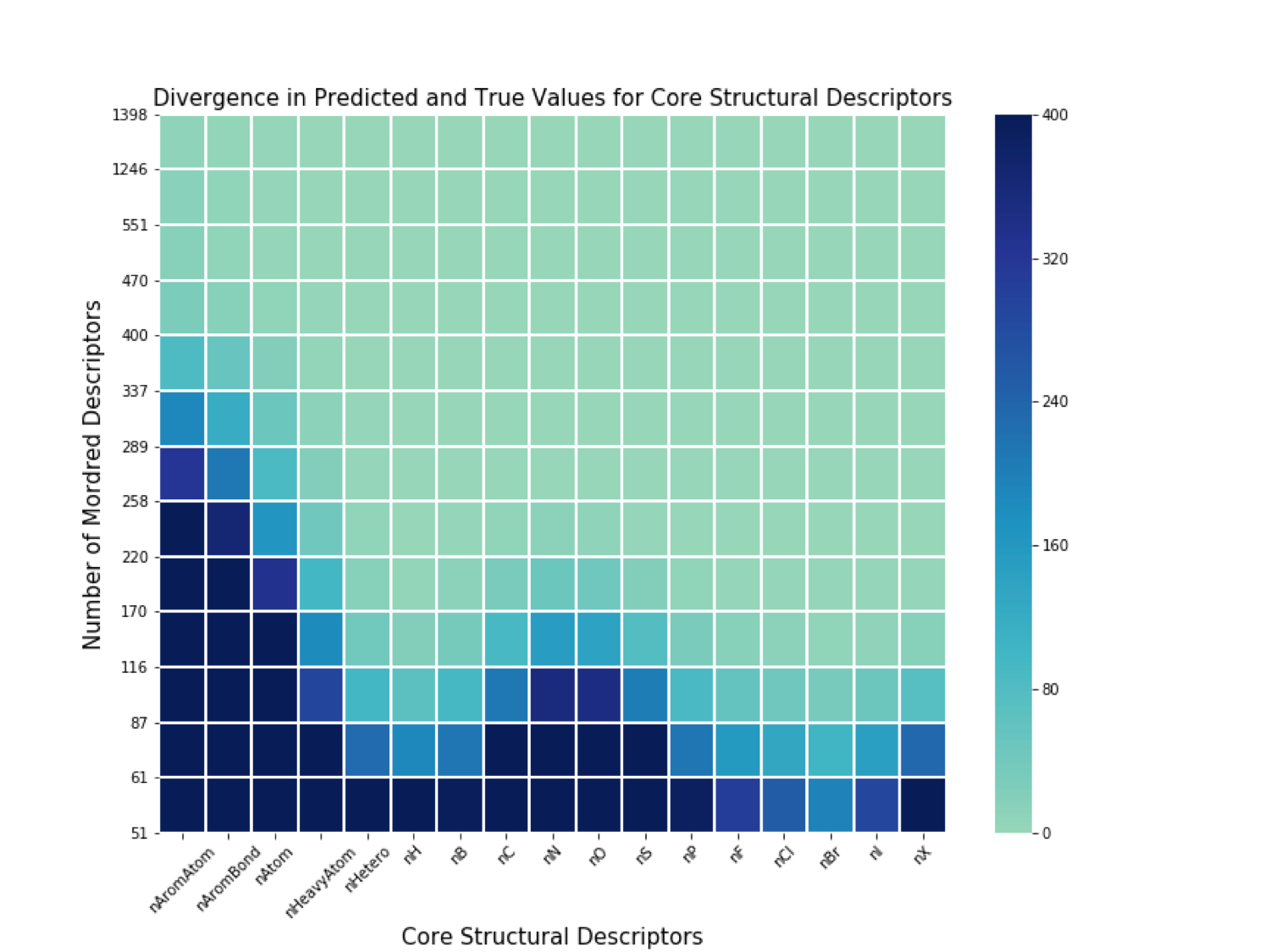
We identified 17 core structural Mordred descriptors that would allow a researcher to effectively deduce the structure of a molecule. Without any of these core structural descriptors, a researcher would not be able to accurately sythesize the structure.
We first attempted data reduction via Principal Component Analysis (PCA) in order to identify the non-core Mordred descriptors that could explain the most variation in our data (i.e. the descriptors that best differentiate the molecules from each other). We did not find any descriptors that significantly affected our principal components.
We then used a multiple output linear regression model. Our model trains using all of the non-core Mordred descriptors in order to predict the value of the core structural descriptors. The model is versatile and is able to integrate other core structural descriptors as predictors, should the user choose to do so.
The Steps
- We cleaned the data. We removed any irregularities from it, and ensured that it consisted only of numerical values.
- We extracted the 17 core structural descriptors.
- We trained a Multiple Output Regressor to predict the 17 core structural descriptors. Using all non-core Mordred descriptors, the model was able to predict of all these descriptors with 100% 5-fold cross-validation accuracy.
- We iteratively thresholded the data, removing the descriptors with the least variance, in order to find the least amount of descriptors required for the model to accurately predict the 17 core structural descriptors.
Results
Using less than around 60 Mordred descriptors, the model poorly predicts all 17 core structural descriptors. A minimum of around 400 Modrerd descriptors are required to identify all 17 core structural descriptors and approximately reverse engineer the molecular structure.
Give it a try!
In order to run our model, do the following:
- Download the Mordred compound sets (1 through 3) and place them in a
data/folder - Open the
src/config.pyfolder and ensure that thedata_pathandresults_pathare correct. - Run the main function in
src/thresholding.py.
Built With
- jupyter-notebook
- python
- scikit-learn

Log in or sign up for Devpost to join the conversation.