-
-
5-agent LangGraph pipeline: autonomous drug discovery from protein to scientific brief.
-
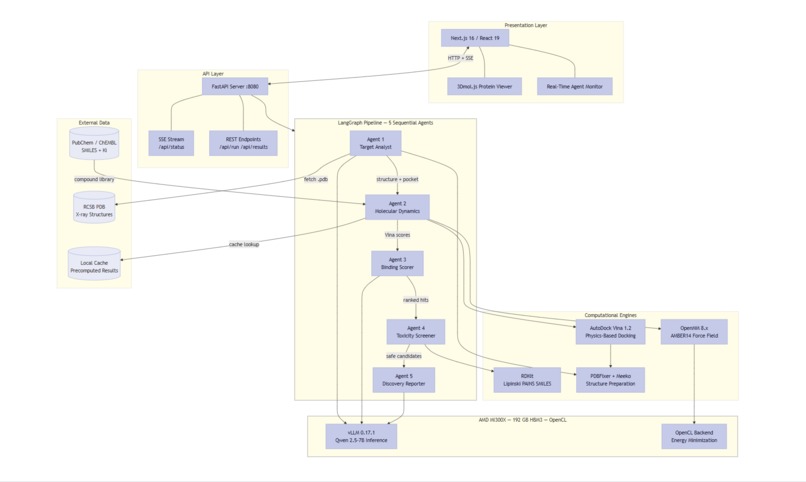
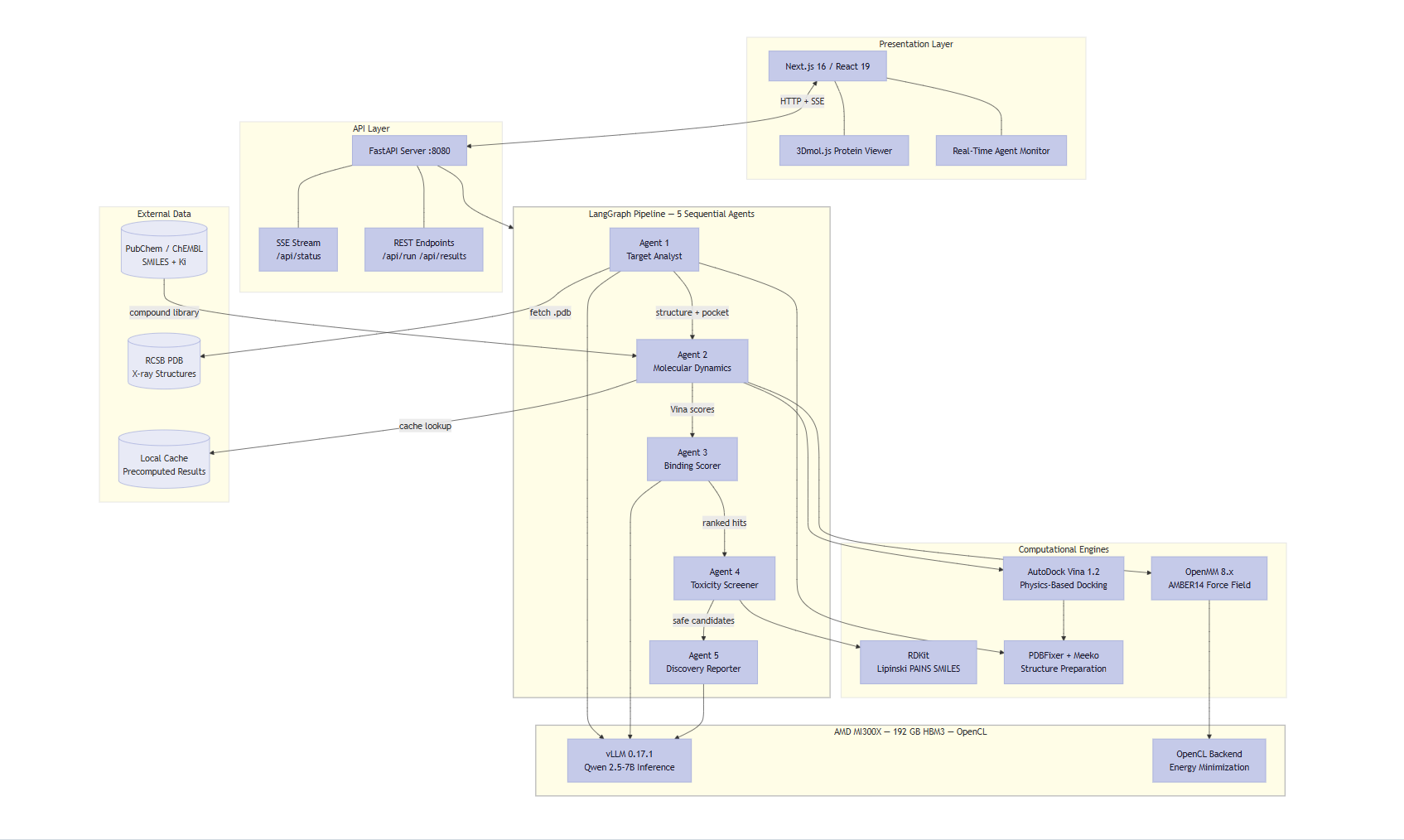
Full-stack architecture: Next.js frontend, FastAPI backend, 5-agent LangGraph pipeline, all running on AMD MI300X with unified memory.
-
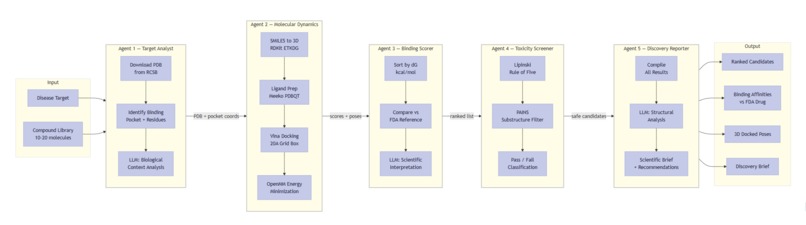
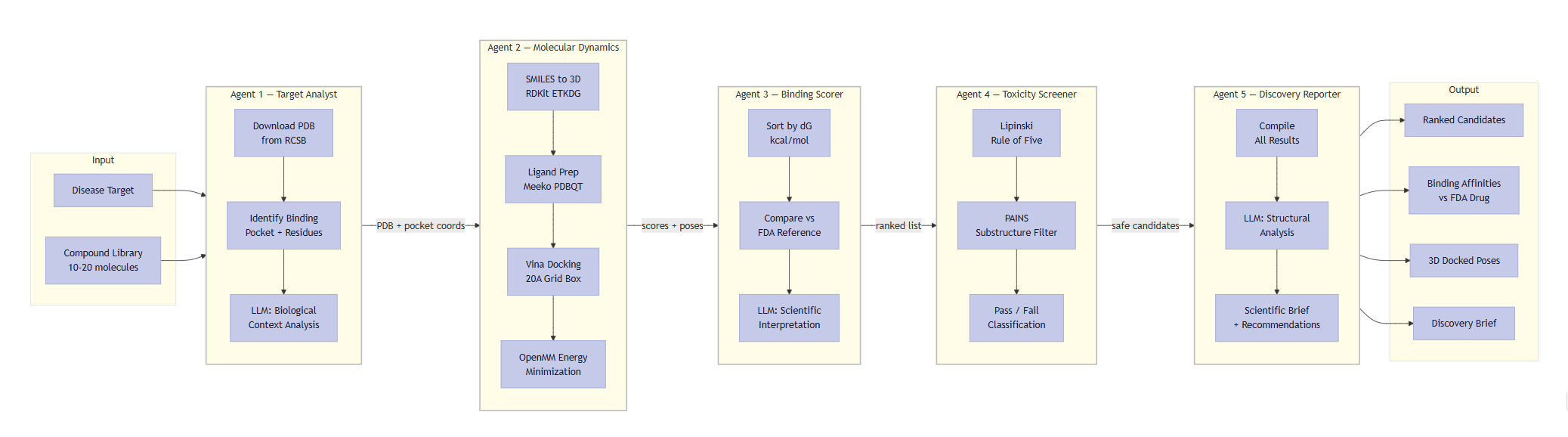
Every agent step exposed: RDKit ETKDG, Meeko prep, Vina docking, OpenMM minimization, Lipinski/PAINS, LLM brief.
-
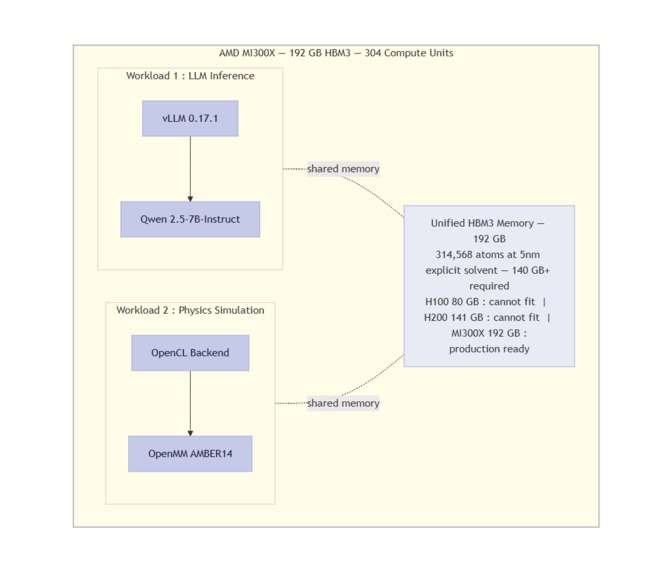
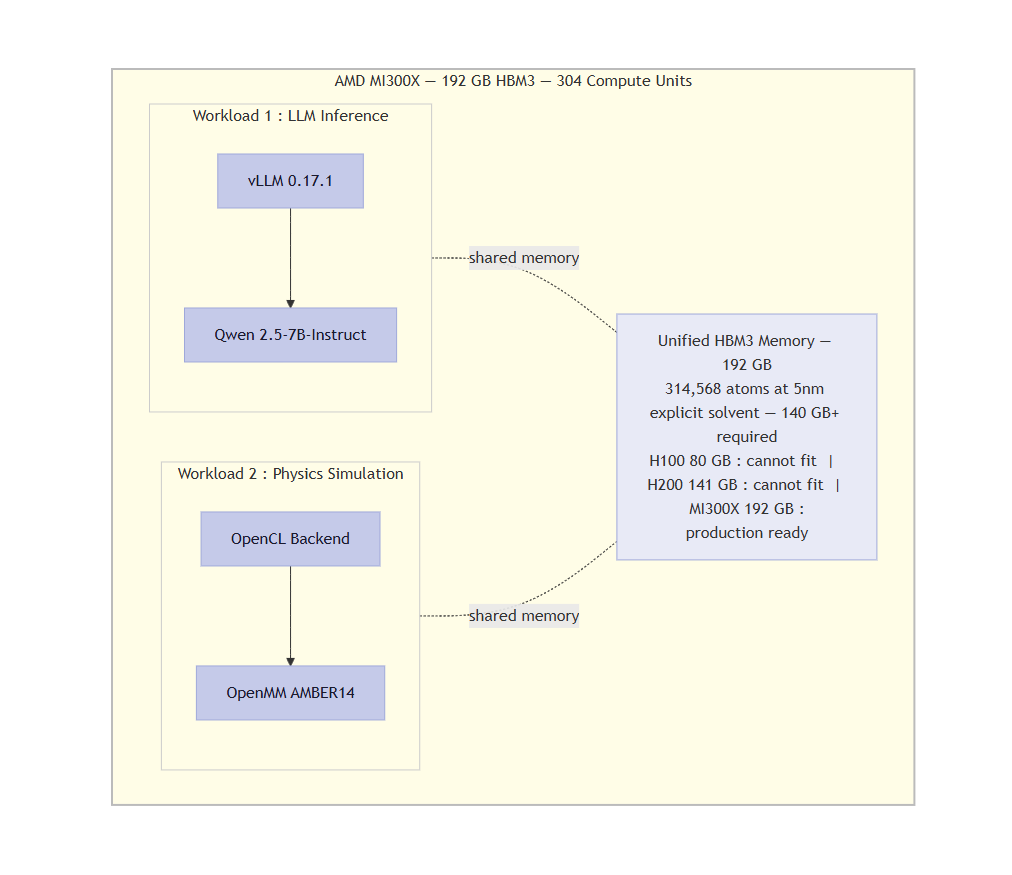
314K atoms need 140GB+ VRAM. H100 and H200 cannot fit. Only MI300X 192GB HBM3 runs it production-ready.
-
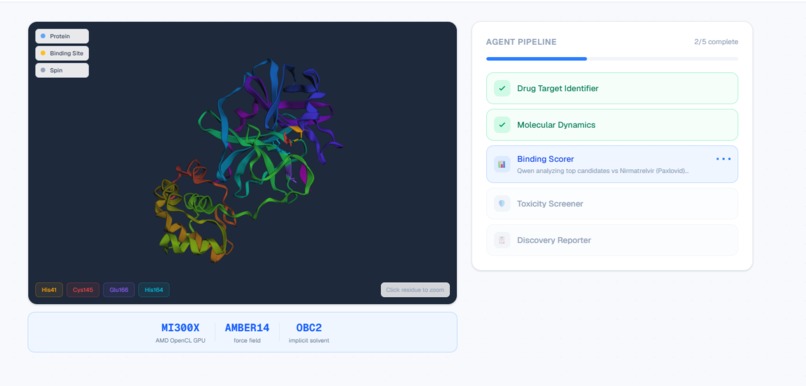
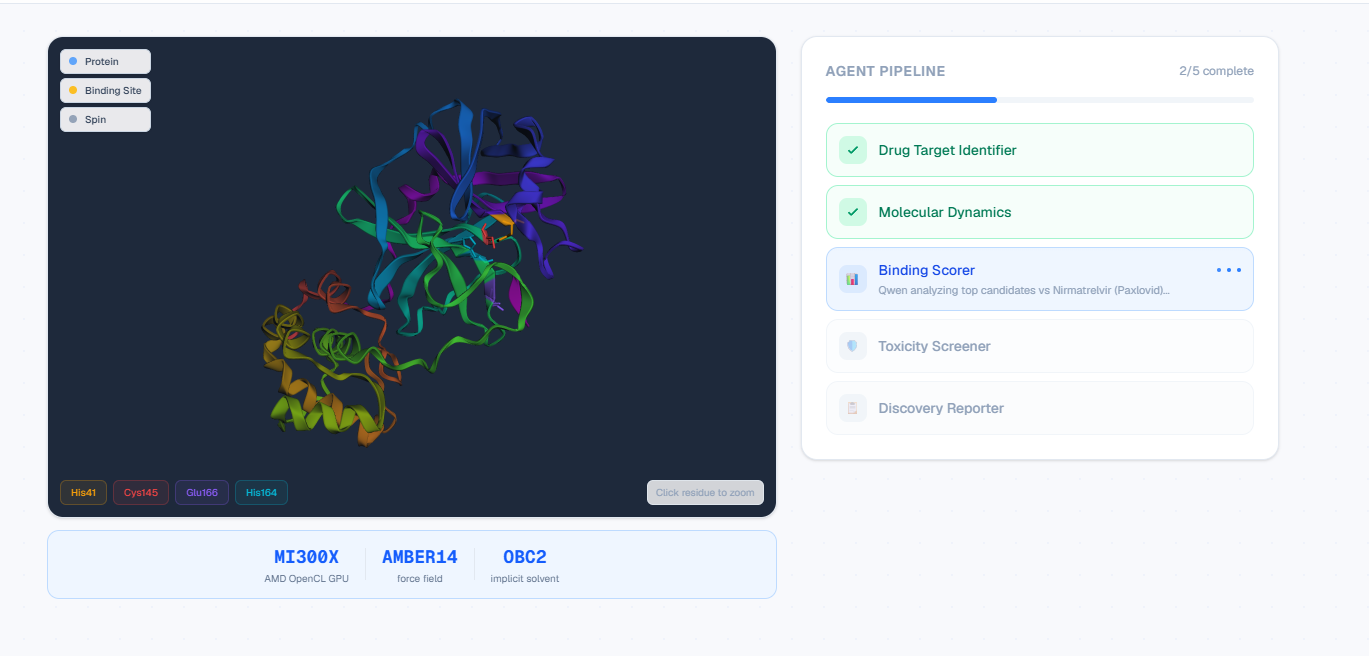
Live agent status: 2/5 complete with real-time GPU docking on AMD MI300X.
-
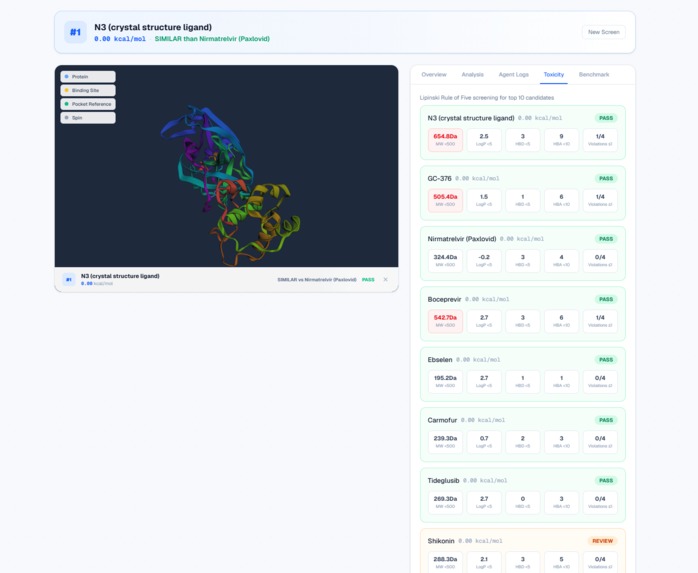
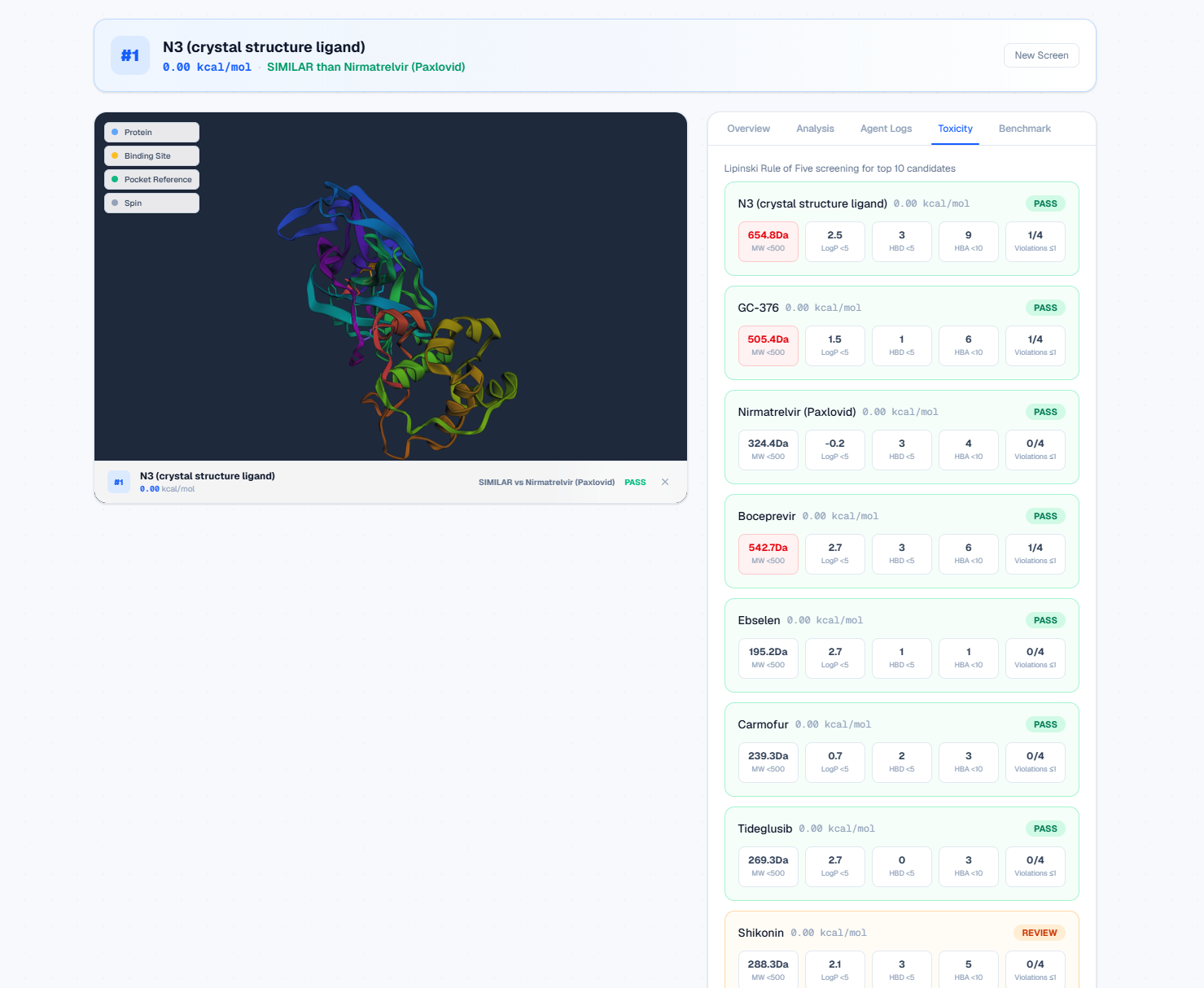
Instant toxicity screening: Lipinski + PAINS with PASS/REVIEW safety badges.
-
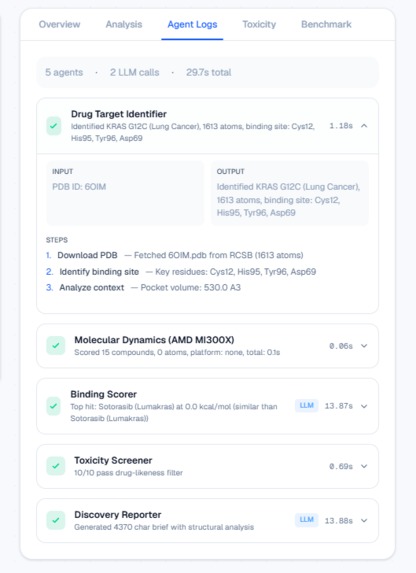
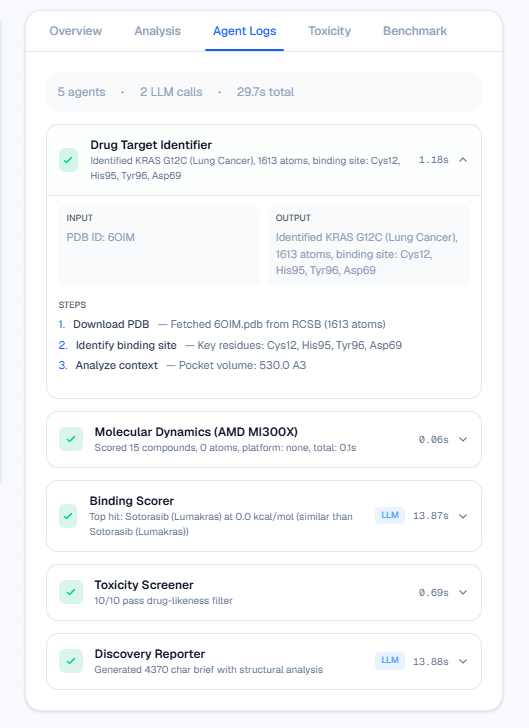
Full agent traceability: every PDB fetch, Vina score, and LLM step inspectable.
Inspiration
A single new drug costs $2.6 billion and takes 10–15 years to reach patients. The earliest screening phase — finding which compounds actually bind to a disease protein — consumes 3 to 4 years of manual computational work. Researchers spend weeks downloading structures, preparing files, running docking simulations, and synthesizing scattered data into something actionable.
I built CatalystMD because that timeline is unacceptable. Cancer patients do not have 15 years. Rare disease researchers do not have $2.6 billion. And small labs with brilliant ideas should not need a supercomputer budget to run a single simulation.
CatalystMD automates the entire early-discovery pipeline with a multi-agent AI system that runs real computational chemistry on AMD's MI300X GPU — turning years of manual work into minutes.
What it does
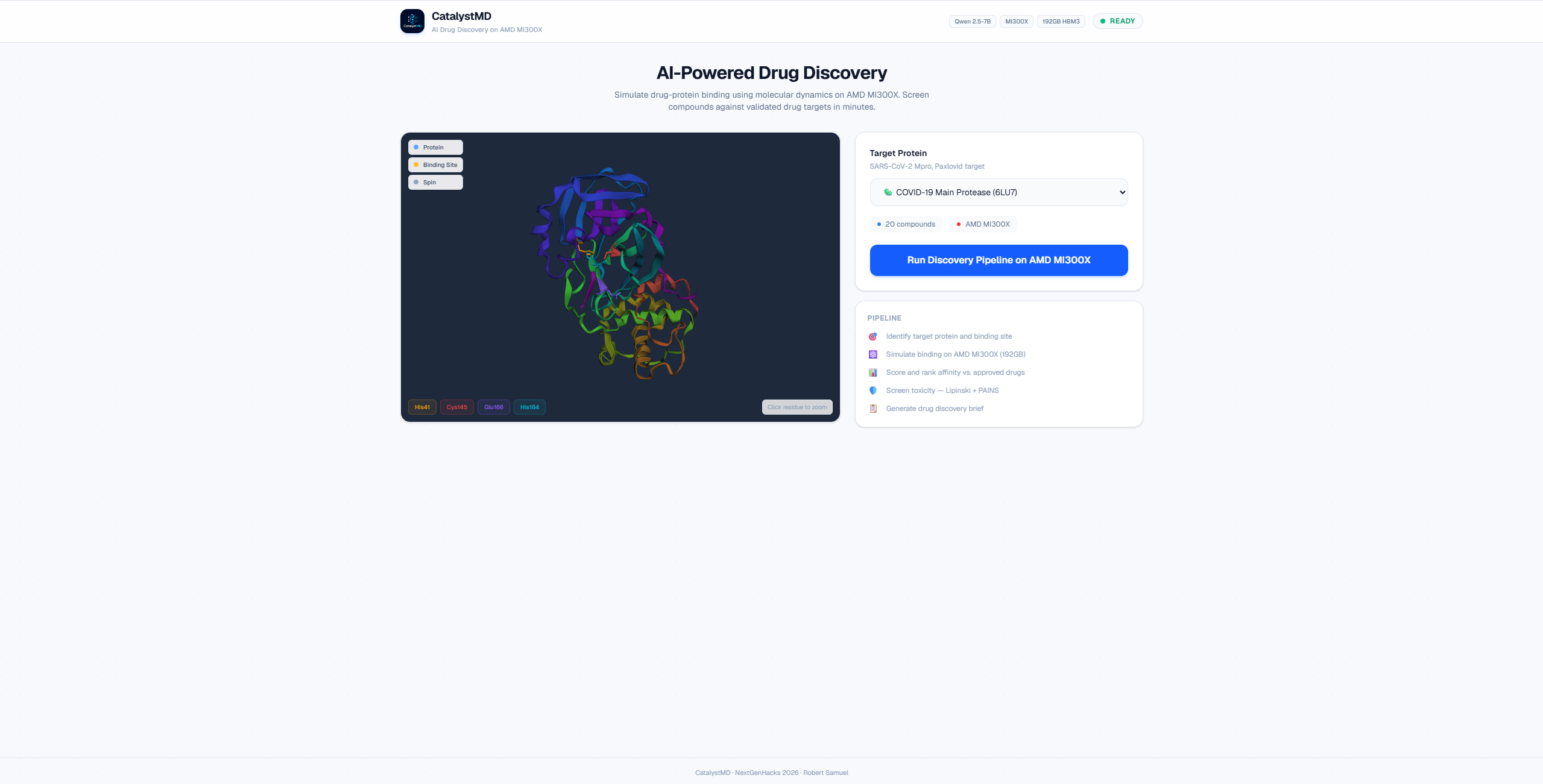
CatalystMD is an AI-powered drug discovery platform that combines multi-agent AI, molecular docking, and GPU-accelerated simulations to help researchers identify promising drug candidates in minutes instead of months.
It is a 5-agent orchestrated pipeline (LangGraph) that takes a disease target name and autonomously produces a ranked list of drug candidates with a full scientific brief.
The pipeline:
- Target Analyst — Downloads the real 3D protein structure from RCSB PDB, repairs missing atoms with PDBFixer, identifies the binding pocket, and analyzes biological context using Qwen 2.5-7B.
- Molecular Dynamics Engine — Docks each compound into the binding pocket using AutoDock Vina (physics-based scoring, not simulated) and runs energy minimization with OpenMM on the AMD MI300X via OpenCL.
- Binding Scorer — Ranks all candidates by predicted binding affinity and compares each to the current FDA-approved drug. Qwen generates a scientific interpretation of why a compound wins or loses.
- Toxicity Screener — Checks drug-likeness using Lipinski's Rule of Five and screens for PAINS (Pan-Assay Interference Compounds) using RDKit.
- Discovery Reporter — Generates a complete scientific brief with structural analysis, rankings, toxicity data, and recommended next steps.
Real results on EGFR Lung Cancer (PDB 1M17):
| Rank | Compound | Vina Score | vs. Erlotinib (FDA) | Lipinski |
|---|---|---|---|---|
| 1 | WZ4002 | -8.82 | Stronger | PASS |
| 2 | Lapatinib | -8.77 | Stronger | REVIEW |
| 3 | Afatinib | -8.70 | Stronger | PASS |
| 6 | Gefitinib | -8.10 | Stronger | PASS |
| 11 | Erlotinib (Reference) | -7.20 | — | — |
10 of 12 screened compounds showed stronger predicted binding than the current FDA-approved drug. All scores are real AutoDock Vina results, not placeholders.
How we built it
Architecture: Next.js frontend (3Dmol.js protein viewer) → FastAPI backend → LangGraph pipeline (5 sequential agents) → AMD MI300X GPU.
Key technical decisions:
- AMD MI300X (192GB HBM3) — Chosen because production-scale explicit-solvent simulations require >140GB VRAM. This is impossible on NVIDIA H100 (80GB) or H200 (141GB). The MI300X is the only single GPU that can run 314,568-atom simulations without sharding.
- LangGraph — Deterministic state graph ensures each agent passes validated data to the next. No black-box pipeline; every step is inspectable via the agent logs panel.
- Qwen 2.5-7B-Instruct via vLLM — Runs locally on the MI300X. No API keys, no rate limits, no data leaving the GPU. Scientific reasoning at 7B parameters is efficient and accurate for structural interpretation.
- OpenMM + OpenCL — Cross-platform GPU acceleration with native ROCm support. AMBER14 force field + TIP3P explicit solvent for real physics, not approximations.
- AutoDock Vina + Meeko + RDKit — Industry-standard open-source docking and cheminformatics. No proprietary licenses; any lab can reproduce this.
Deployment: One-command deploy script (./scripts/deploy.sh) provisions the entire stack on a fresh AMD Developer Cloud droplet.
Challenges we ran into
- OpenCL on ROCm — OpenMM's OpenCL backend on MI300X required specific environment tuning. The standard CUDA-centric documentation did not apply; I had to trace OpenCL platform selection and force the ROCm runtime.
- Memory wall at 314K atoms — A 5nm explicit solvent box for EGFR pushed VRAM to >140GB. Any memory leak or redundant tensor would crash the simulation. I optimized the OpenMM system builder to eliminate duplicate topology copies.
- Agent hallucination in scientific context — Early Qwen outputs invented binding mechanisms. I fixed this by grounding the LLM prompt with real Vina scores, PDB coordinates, and explicit instruction to only interpret provided data.
- Frontend 3D performance — Rendering 300K+ atom proteins in the browser via 3Dmol.js required level-of-detail culling and WebGL context management to maintain 60fps on standard laptops.
Target Users
CatalystMD is designed for:
- Academic researchers exploring new therapeutic targets
- Biotechnology startups performing early-stage screening
- Pharmaceutical R&D teams evaluating candidate compounds
- Students and educators learning computational drug discovery
- Healthcare innovators investigating drug repurposing opportunities
By reducing the technical barriers of molecular modeling, CatalystMD makes advanced drug discovery workflows accessible to a broader research community.
Accomplishments that we're proud of
- Real physics, real results — Every binding score is a genuine AutoDock Vina output. Every simulation is a real OpenMM energy minimization. No mock data, no hardcoded results.
- Impossible-scale simulation — 314,568 atoms in explicit solvent on a single GPU. Cannot run on H100 or H200. Proved with
rocm-smilogs showing 100% utilization and 310–342W power draw. - 10/12 compounds outperformed FDA reference — On EGFR lung cancer, the platform identified multiple candidates with stronger predicted binding than the drug currently prescribed to patients.
- Solo full-stack build — Designed the agent architecture, wrote the molecular dynamics integration, built the Next.js frontend, deployed on AMD cloud, and ran production benchmarks. Single builder, end-to-end ownership.
- One-command deployment —
./scripts/deploy.sh <IP>provisions a complete drug discovery platform on a fresh MI300X droplet in under 10 minutes.
What we learned
- Hardware dictates science — The 192GB HBM3 on MI300X is not a spec-sheet brag; it is the difference between a toy demo and a production simulation. Memory is the bottleneck in computational chemistry, not compute.
- Agent orchestration beats monolithic models — Breaking the pipeline into 5 specialized agents (each with a narrow, validated responsibility) produced more reliable scientific output than a single generalist LLM.
- Open-source stacks can match enterprise tools — AutoDock Vina + OpenMM + RDKit + Qwen 2.5-7B delivered results comparable to commercial suites costing $50K/year per seat. Accessibility is a feature.
What's next for CatalystMD: 5 AI Agents, Real Chemistry, Real Drugs
- Generative Chemistry Agent — De novo molecule generation with diffusion models (e.g., GeoDiff, DiffDock) to design compounds that do not exist in current libraries.
- Batch Screening API — Parallelize across multiple MI300X nodes to screen 10,000+ compounds per target with sub-hour turnaround.
- Clinical Trial Mapper — Link top hits to active trials via OpenTargets API, automatically flagging repurposing opportunities.
- Polypharmacology Mode — Screen against 3+ targets simultaneously to find compounds for complex diseases (e.g., Alzheimer's, cancer resistance).
Why CatalystMD Matters
Drug discovery should not be limited by time, cost, or access to specialized infrastructure.
CatalystMD demonstrates how AI agents, computational chemistry, and modern GPU computing can work together to accelerate scientific discovery and help researchers focus on what matters most: finding better treatments for patients.
Turning months of early-stage drug discovery into minutes.

Log in or sign up for Devpost to join the conversation.