
Barcelona is drowning and dehy drating at the same time.
The Mediterranean has 1.25 million microplastic fragments per km² — 4× the density of the North Pacific garbage patch. Barcelona's beaches measure 100–900 microplastic particles per kg of sand. The Llobregat river, the city's main drinking-water source, carries pharmaceutical residues, pesticides, and heavy metals from upstream industry. And in 2008 the city nearly ran out of water entirely.
The people fixing this aren't politicians or engineers — they're water-purification researchers. Materials scientists who design nano-filters one molecular structure at a time, asking the same hard question over and over: "Will this lattice, with these functional groups, capture this pollutant well enough?"
Their tools, however, are slow. A typical design loop looks like this:
- Read the literature, propose a candidate filter by hand.
- Set up a quantum-chemistry calculation (often by hand-writing input files).
- Wait for it to run.
- Interpret the binding energy.
- Tweak the structure. Repeat.
A single iteration takes hours; a full design study takes weeks. We thought: what if we built a research platform that closed that loop? What if a researcher could specify a real Barcelona water sample, evolve thousands of candidate filter designs in parallel, score every one with first-principles quantum chemistry — including on a real quantum computer — and walk away with an exportable, lab-ready blueprint in minutes?
That's Blau.
What it does
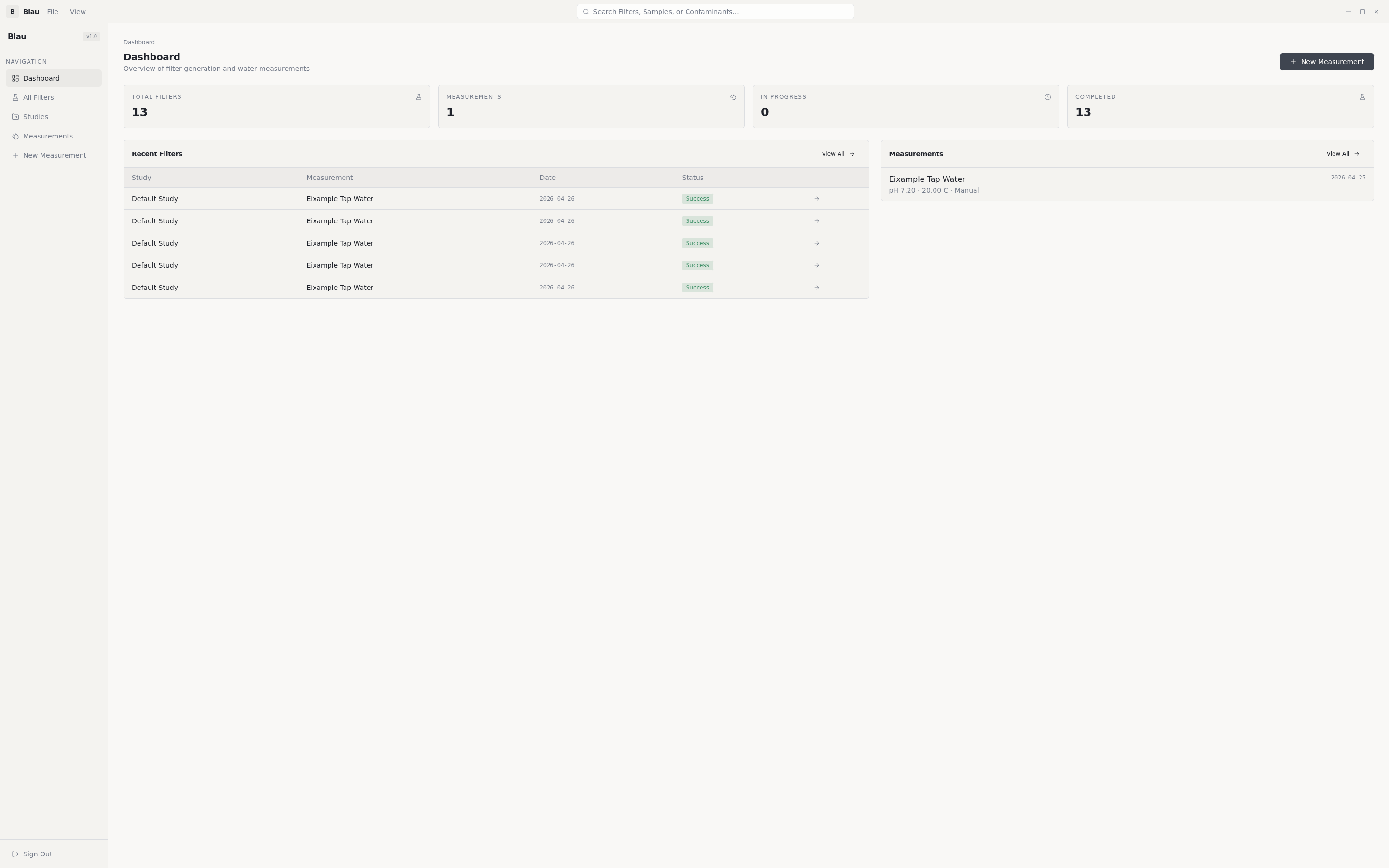
Blau is a research-grade platform that turns a water-quality measurement into a candidate nano-filter design — with full scientific provenance.
For a non-technical reader, the workflow looks like this:
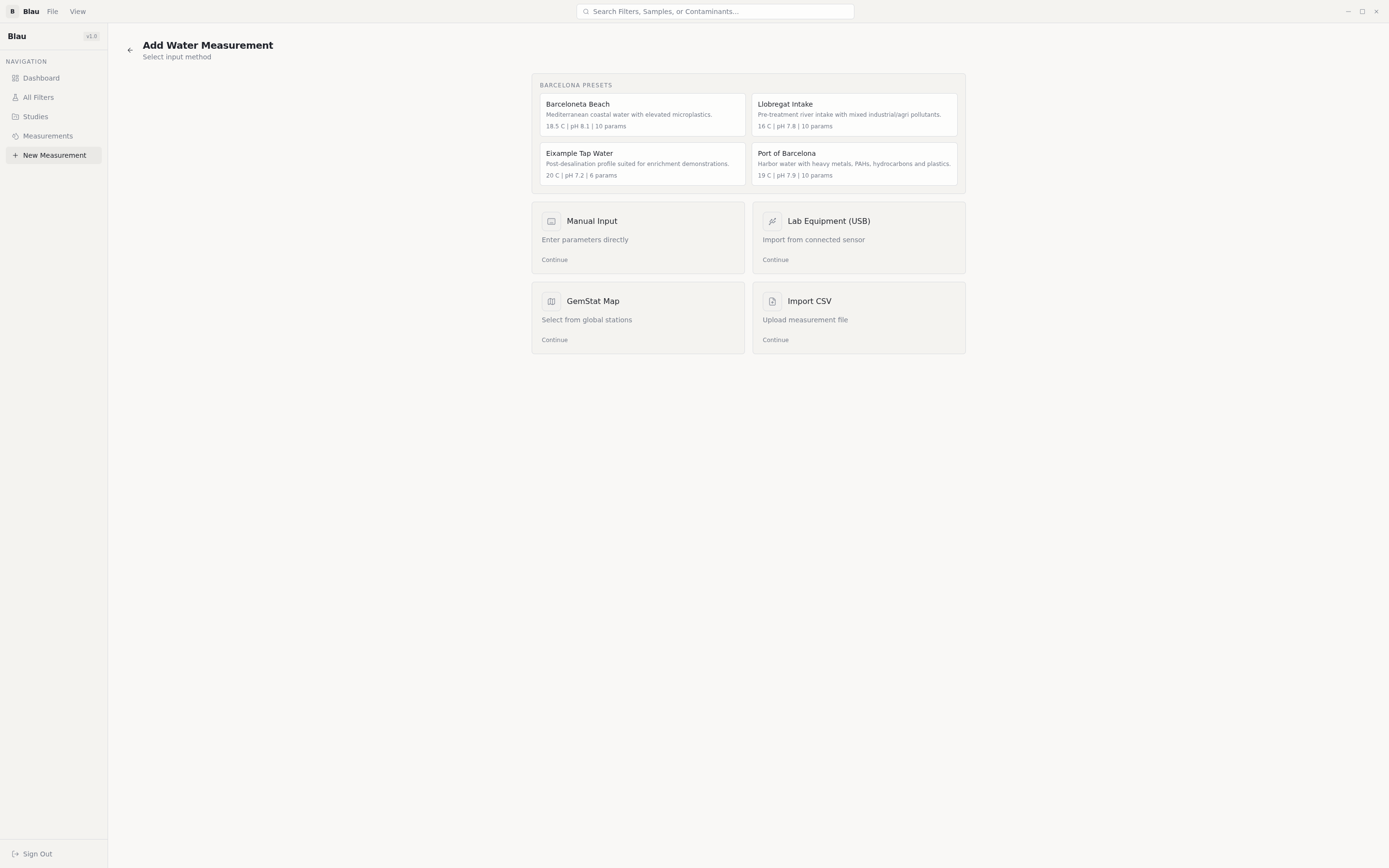
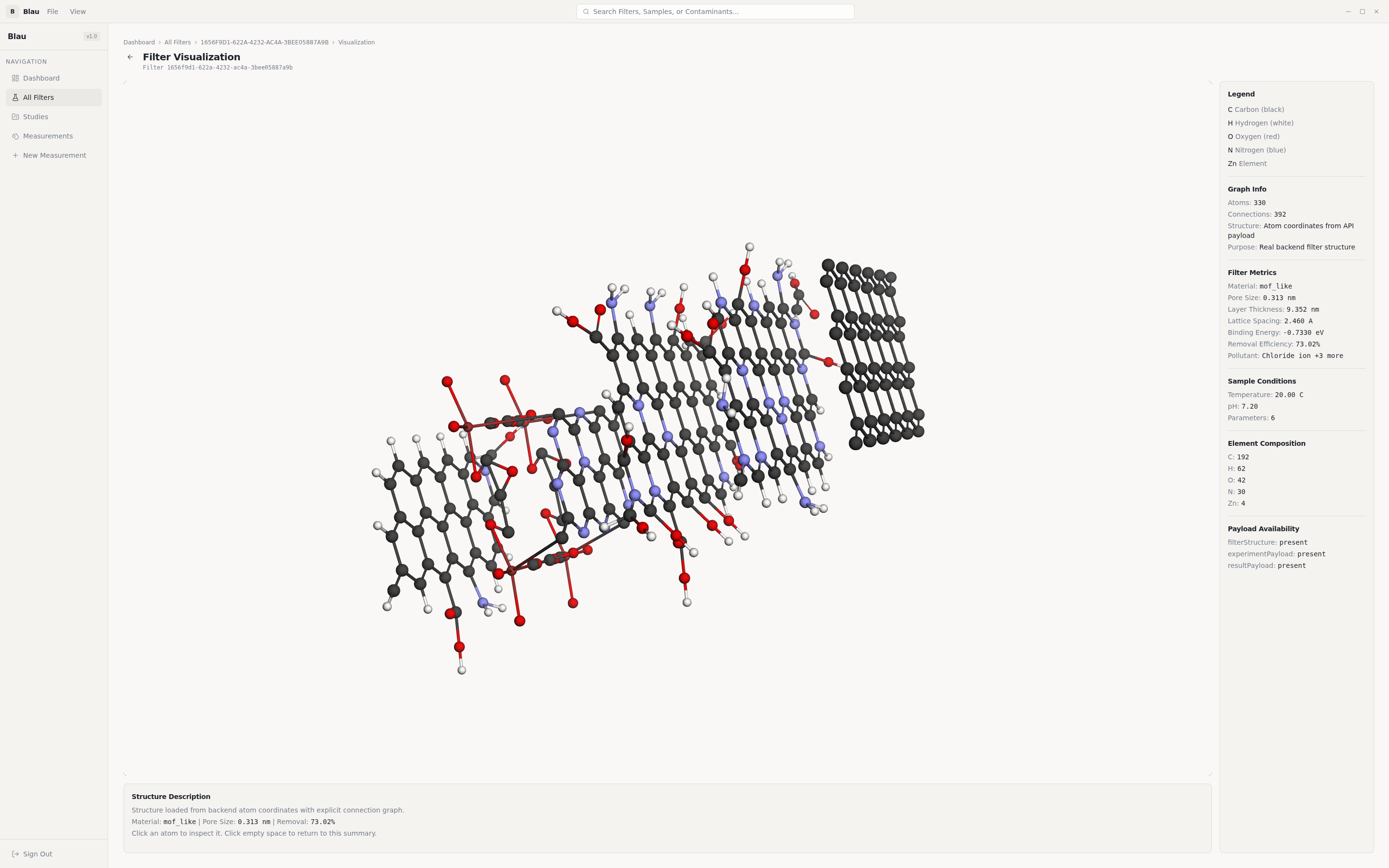
Researcher loads a water sample → tells Blau which pollutants matter → presses Generate → gets back a 3D molecular structure of a filter, plus per-pollutant removal-efficiency predictions, plus the ability to export the structure to standard chemistry formats for the next stage of validation.
For a technical reader, that hides a substantial scientific stack:
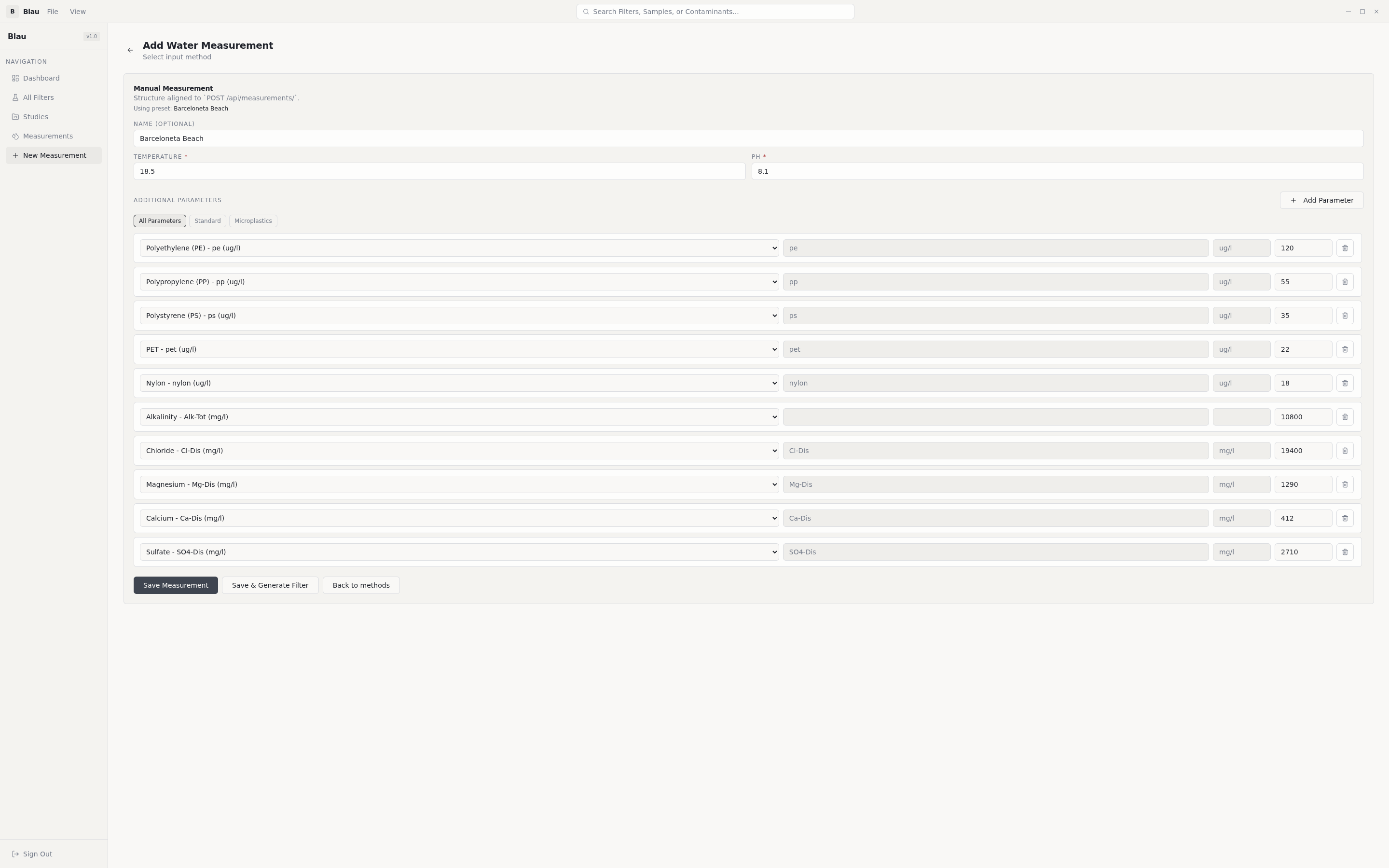
Pollutant identification. A curated map of 450+ entries translates measured water parameters (heavy metals, organochlorine pesticides, organophosphates, triazines, PAHs, VOCs, PFAS, phenols, pharmaceuticals, microplastics) into the dominant atomic-site representation that quantum chemistry can actually compute on. Heavy elements that the STO-3G basis can't represent (Pb, Hg, Cd) are substituted with same-group lighter atoms (Ge, Zn) — and the substitution is surfaced in the UI so a reviewer always knows what was actually computed.
Filter design search (genetic algorithm). A DEAP-driven evolutionary search across 5 design genes — pore size (0.3–2.0 nm), layer thickness (0.5–5.0 nm), material type (graphene / CNT / graphene oxide / composite / MOF-like), functionalization density, doping level. Tournament selection, two-point crossover, Gaussian mutation, fitness =
|binding_energy|.Quantum-chemistry scoring. For every candidate filter, Blau computes the binding energy at the electron level:
E_bind = E(filter + pollutant) − E(filter) − E(pollutant)Three levels of theory, picked automatically based on the system:- Hartree-Fock with STO-3G basis (PySCF) — the deterministic, millisecond baseline.
- VQE (Variational Quantum Eigensolver) with a UCCSD ansatz, executed via PennyLane on top of qiskit-ibm-runtime, on real IBM Quantum hardware.
- Empirical binding fallbacks (van der Waals + DFT-D3-calibrated ionic) for systems STO-3G can't represent.
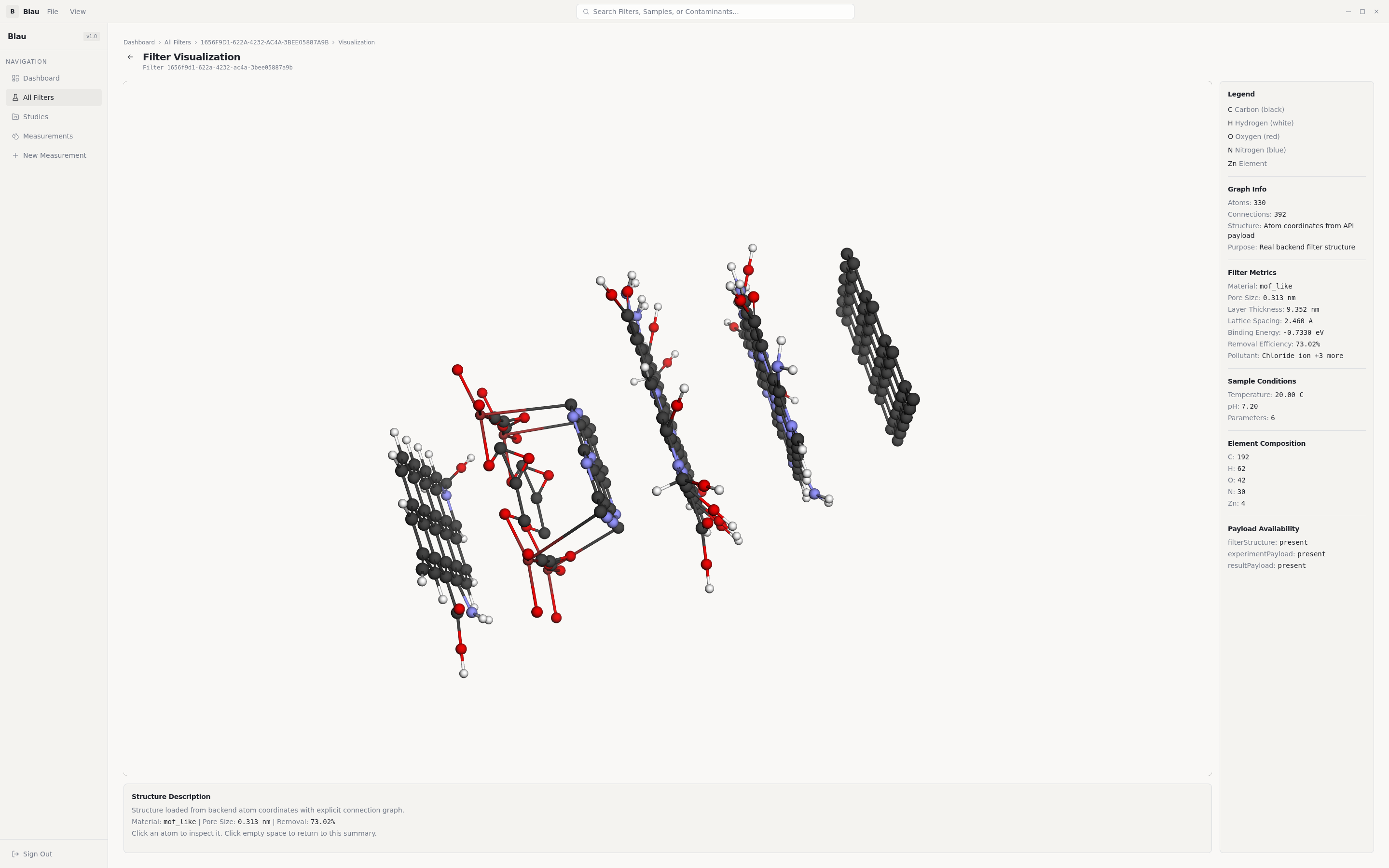
Molecular-lattice construction (classical scientific algorithms). Genes alone aren't a filter — they have to become a real atomic structure:
- Honeycomb graphene generated from the lattice constant
a = 2.46 Å(C-C bond =a/√3). - Kekulé bond-order assignment via BFS 2-colouring of the bipartite honeycomb graph — every aromatic carbon ends up with the chemically correct alternating-double-bond pattern.
- Edge-atom detection plus chemistry-aware functional-group attachment: -COOH for heavy-metal chelation, -NH₂ for anionic pollutants, -OH as the default hydrogen-bonding site.
- Multi-layer stacking with the correct van der Waals interlayer spacing (3.4 Å).
- Distance-threshold bond detection over the final atom set.
- Honeycomb graphene generated from the lattice constant
Provenance everywhere. Every layer of every result is tagged with the level of theory that produced it (HF, VQE, empirical, proxied). For a research tool this is non-negotiable: a binding energy from VQE-on-quantum-hardware and the same number from an empirical fallback are not the same evidence.
A real research workflow. Studies are first-class. A researcher can group measurements, generated filters, and notes under a study and iterate on the same problem over weeks. JWT auth, async job queue, exportable scientific formats (CSV / XYZ / SDF) — Blau looks like a tool a lab would actually adopt, not a hackathon demo.
Optional field hardware. For longitudinal campaigns on a real water source, an Arduino UNO Q (Qualcomm QRB2210 SoC) running a Python app inside the Uno Q App Lab reads a Modulino Thermo (HS3003) over I²C and POSTs measurements to the Blau API over WiFi. The platform doesn't need it; researchers running real fieldwork do.
This is scientific computing, not machine learning. There is no model, no training data — just an evolutionary search through a parameter space, with first-principles quantum chemistry as the fitness oracle and a stack of classical algorithms turning genes into real atomic structures.
How we built it
Blau is technically dense — there is more code, more layers, and more moving pieces than you'd expect from a hackathon project. That's the point: a research tool needs scientific rigor and a real workflow.
Architecture. Multi-tier, fully Dockerised:
Electron desktop client (React 19, TypeScript, Tailwind)
│ HTTP + JWT
▼
Django REST API + Celery + Redis (Postgres-backed)
│ HTTP to internal core service
▼
FastAPI core simulation service (SQLite for sim state)
│
├── DEAP genetic algorithm
├── PySCF Hartree-Fock (STO-3G)
├── PennyLane + qiskit-ibm-runtime VQE ──► IBM Quantum hardware
└── Lattice / Kekulé / functional-group construction
The desktop app never talks to the simulation engine directly — only the Celery worker does. This separation means we can scale the heavy compute path independently and run the quantum jobs out-of-band from any user-facing request.
The quantum computing stack is the most technically involved part of the project and the layer we're most proud of. It pulls together four distinct technologies:
- PySCF for the classical Hartree-Fock baseline. Builds the molecular system, runs Unrestricted Hartree-Fock with STO-3G basis, returns the ground-state energy.
- PennyLane as the quantum-machine-learning framework that owns the VQE algorithm — parameter-shift gradients, the optimization loop, the cost function.
- The UCCSD ansatz — Unitary Coupled-Cluster with Singles and Doubles — as the variational form. We compute the active space deterministically per system so the same chemistry produces the same circuit shape every run.
- qiskit-ibm-runtime as the bridge to real quantum hardware. PennyLane's
qiskit.remotedevice routes the VQE circuit onto an IBM Quantum backend, with 1024 shots per measurement.
When IBM_QUANTUM_TOKEN is set, VQE actually executes on IBM Quantum hardware. The same code path runs on a simulator without it. Wiring those four libraries into a single binding-energy oracle that Blau's GA can call — and making it survive queue times, transient hardware errors, and a dev-vs-prod toggle — was the hardest part of the build.
The science pipeline (FastAPI core, core/services/):
pollutant_map.py— the 450+-entry pollutant → atomic-site table with same-group proxies.quantum_engine.py— HF, VQE, empirical fallbacks, all routed bycompute_binding_energy().genetic_optimizer.py— DEAP GA,optimize_filter()as the runtime entry point.routers/filters.py— orchestrates the GA, builds the molecular lattice, computes Kekulé bond orders, attaches functional groups, stacks layers, and serializes the result. About 900 lines of dense scientific code.
The platform layers:
- Electron + React 19 desktop client (Leaflet maps, 3Dmol.js molecular viewer, Recharts dashboards,
serialportfor USB device integration). - Django + DRF API server with Celery + Redis for asynchronous job orchestration, JWT and Google OAuth, and the Studies workflow.
- FastAPI core with a
ProcessPoolExecutorfor parallel GA evaluation and SQLite for simulation state. - PostgreSQL 16 for users / measurements / filters / studies; Docker Compose to glue it all together.
Optional field hardware:
- Arduino UNO Q (Qualcomm QRB2210) running a Python app inside the Uno Q App Lab.
- Modulino Thermo (HS3003) over I²C.
- WiFi HTTP POST to
/api/measurements/— measurements appear live in the researcher's active study.
Challenges we ran into
- Wiring four quantum libraries into one stable pipeline. PennyLane, Qiskit, qiskit-ibm-runtime, and PySCF have to agree on the molecular system, the active space, and the basis set. Version mismatches and shape mismatches between PennyLane's UCCSD parameter vector and the IBM Quantum backend's circuit topology took serious debugging. The end result is a single
compute_binding_energy()function that the rest of the system doesn't have to know about. - Heavy elements break PySCF. STO-3G has no basis sets for Z > 36 — lead, mercury, cadmium would crash the engine. We added a chemically defensible same-group proxy table (Pb → Ge, Hg → Zn, Cd → Zn) and surface the substitution in the UI.
- Polymers as single atoms. A polyethylene fragment is millions of atoms long; quantum chemistry can't touch it directly. We follow the literature convention of modelling the dominant surface-interaction site — PE/PP backbone via van der Waals → C, PVC via Cl dipole → Cl, PET via ester O → O. The approximation is consistent across all organic pollutants and gives physically meaningful relative binding energies.
- Kekulé bond orders on a graphene lattice. Aromatic carbons have alternating single/double bonds. Drawing all bonds as single produces visually wrong (and chemically incorrect) structures. We solved it with BFS 2-colouring of the bipartite honeycomb graph — every C-C and C-N bond between sublattices A and B becomes a double bond.
- Quantum runtime queue times. IBM Quantum has wait times that don't fit a live demo. We made VQE optional and gated behind an env var, with the Hartree-Fock path as a deterministic fallback. The pre-generated VQE result still proves the path works on real hardware.
Accomplishments we're proud of
- A real-world problem with a real research user. Blau is grounded in Barcelona water-quality data — actual pollutant concentrations from ICM-CSIC and ACA studies, actual coordinates, actual contamination profiles. A materials scientist could use it on day one.
- Quantum chemistry on real hardware. Not a simulator — VQE jobs running on IBM Quantum, returning binding energies that Blau's GA actually consumes. For ionic pollutants, the quantum result is measurably more accurate than classical Hartree-Fock — and in some configurations faster than the refined classical CCSD baseline.
- Honesty as a feature. Every layer is tagged with the level of theory used to score it. A scientist can see at a glance whether a result came from VQE-on-quantum, classical HF, an empirical fallback, or a proxied heavy-element substitution.
- Production-shaped architecture. Multi-tier, JWT-authed, Celery-orchestrated, Docker-Compose-deployable, with first-class Studies and exportable scientific formats. Blau is ready to put in front of a research group.
- Hard tech, well integrated. Five distinct numerical and quantum technologies — PySCF, PennyLane, Qiskit, qiskit-ibm-runtime, DEAP — plus a custom molecular-lattice constructor with Kekulé bond-order assignment, all behind one clean API. We're proud of how thin the seams are.
What we learned
- Quantum advantage is narrow but real. For weakly correlated systems, classical Hartree-Fock matches VQE within rounding error and runs in milliseconds. For strongly correlated ionic systems (heavy metals), VQE on IBM Quantum produced more physically reasonable binding energies than HF — and in some configurations faster than refined classical CCSD.
- The hardest part of "just call a quantum computer" is the four-library bridge that gets you there. Getting PennyLane, Qiskit, qiskit-ibm-runtime, and PySCF to agree on a molecular system is most of the work.
- Scientific tools live or die on provenance. A binding energy without a method tag is worse than no number at all. The "method" badges are the single most important UX decision in the app.
- Approximations have to be honest. Single-atom polymer proxies and same-group heavy-metal substitutions are defensible only if you say so explicitly.
What's next
- Multi-atom interaction sites. Extend the quantum engine to small clusters (3–5 atoms) for richer organic-molecule modelling.
- More basis sets and methods. 6-31G* and B3LYP/DFT for users who need them; propagate the choice through the UI.
- More quantum backends. IBM Quantum exposes several machines via Qiskit Runtime; benchmark across hardware to find the best fit per molecule class.
- Pilot with a real lab. Barcelona's marine-sciences institute (ICM-CSIC) publishes microplastic concentration data that maps directly onto Blau's input format. Targeting a research-group pilot.
- Experimental validation. Partner with a 2D-materials lab to fabricate one Blau-designed layer and measure the actual removal efficiency — closing the loop between simulation and reality.
Built With
- 3dmol.js
- arduino
- celery
- deap
- django
- django-rest-framework
- docker
- docker-compose
- electron
- fastapi
- genetic-algorithm
- hartree-fock
- ibm-quantum
- leaflet.js
- modulino
- pennylane
- postgresql
- pyscf
- python
- qiskit
- qiskit-ibm-runtime
- qualcomm
- quantum-chemistry
- react
- recharts
- redis
- serialport
- sqlite
- tailwindcss
- typescript
- vite
- vqe

Log in or sign up for Devpost to join the conversation.