-
-
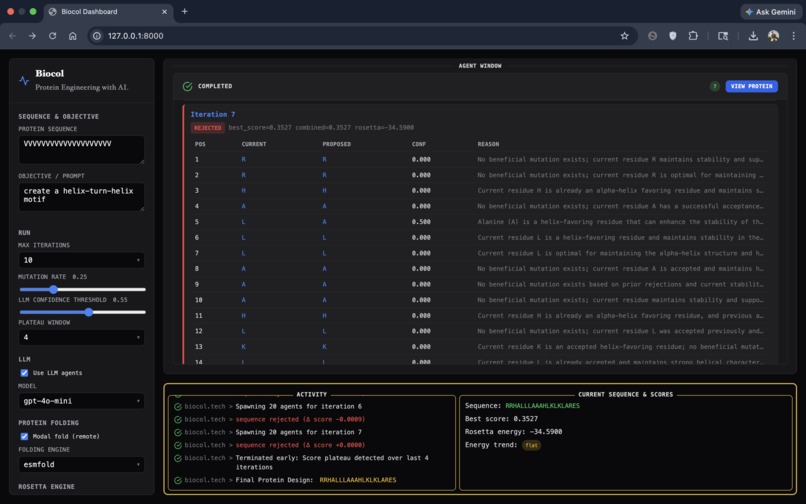
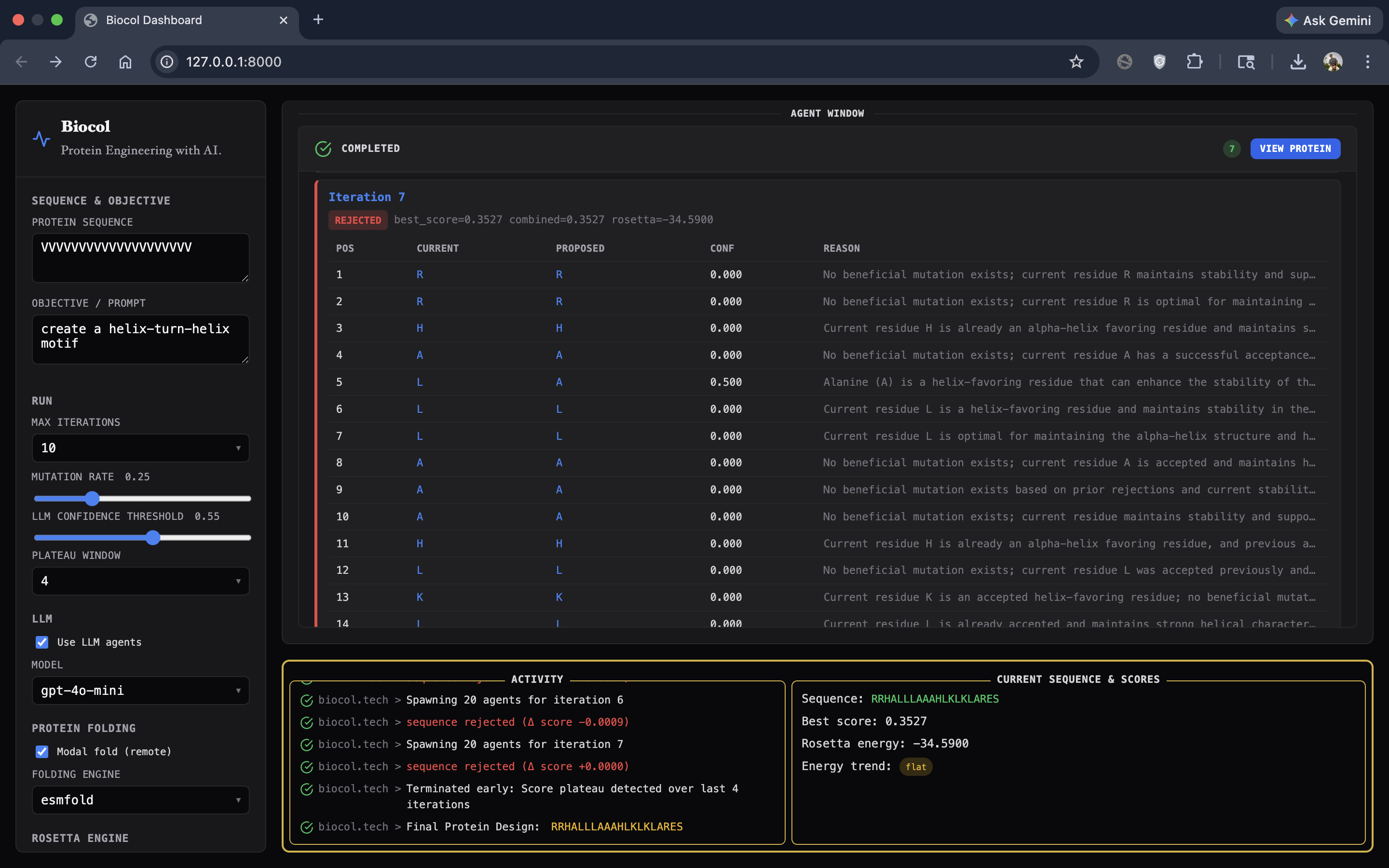
BioCol's user interface - shows a successful protein design from a starting sequence of amino acids.
-
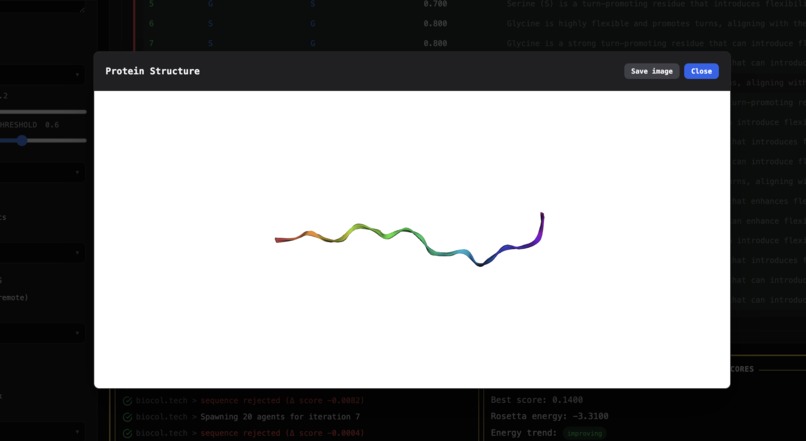
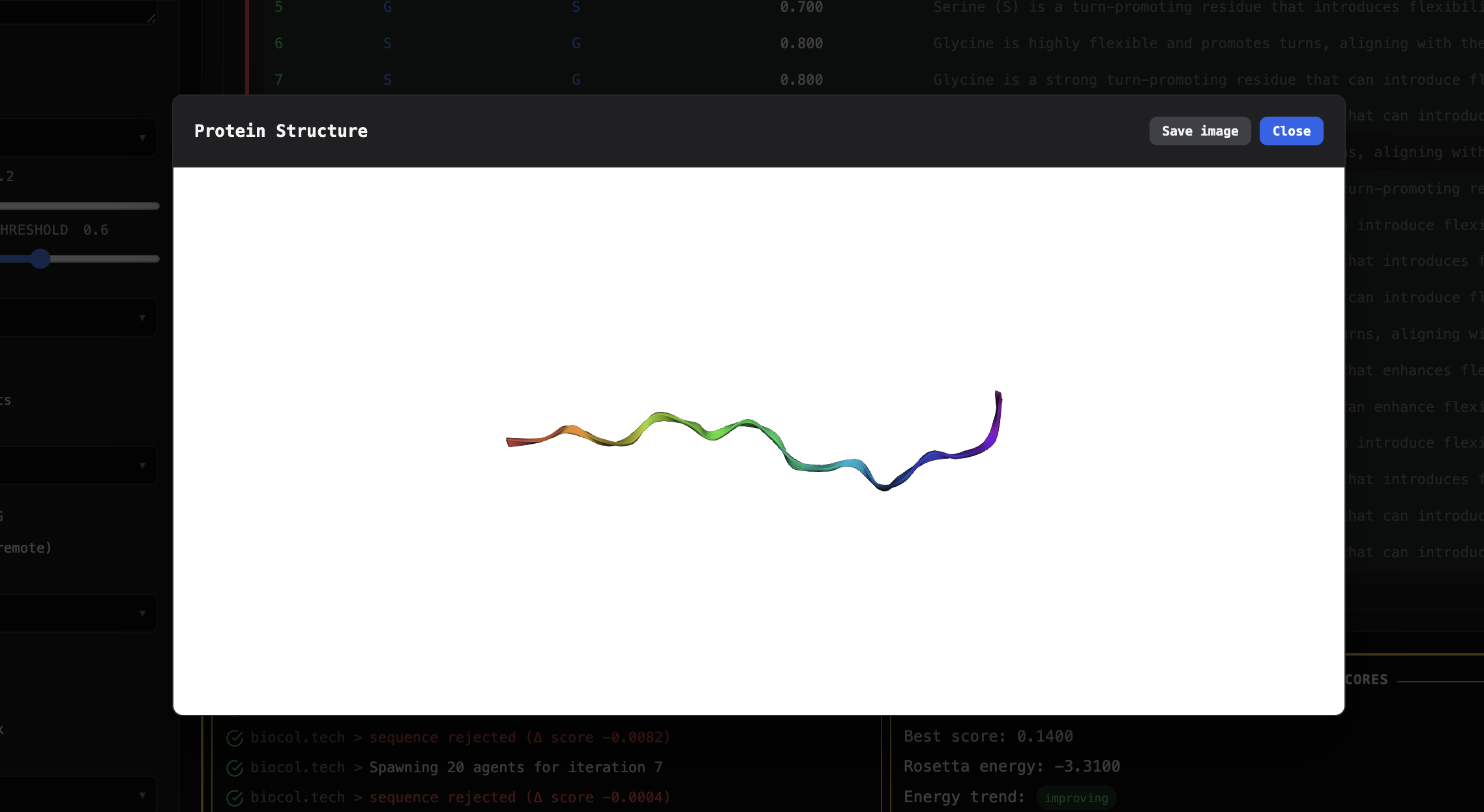
A 3D rendered image of the proposed protein design.
-
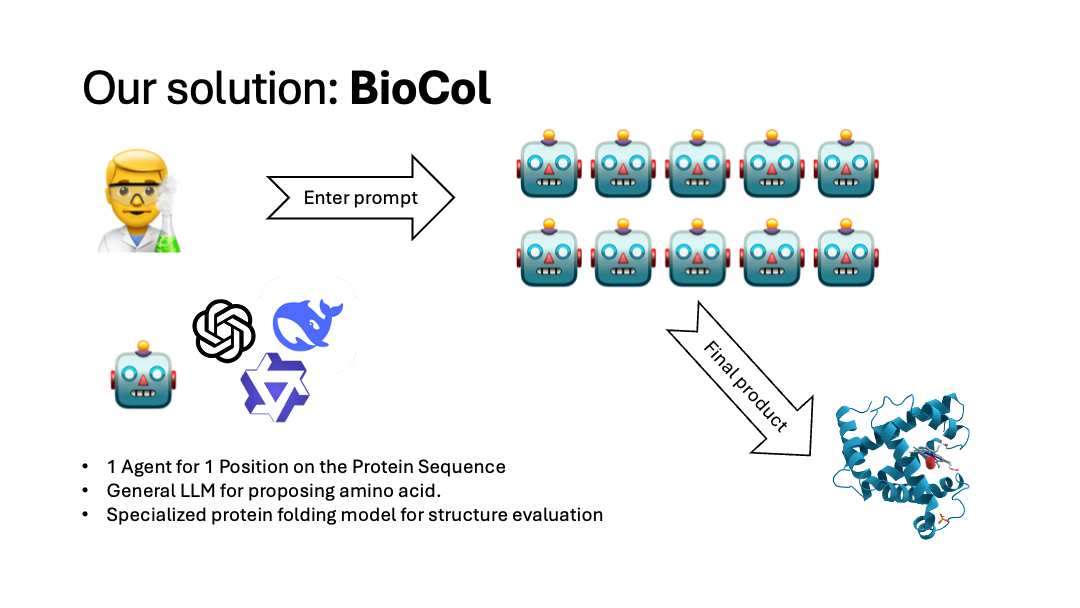
A swarm of AI Agents.
-
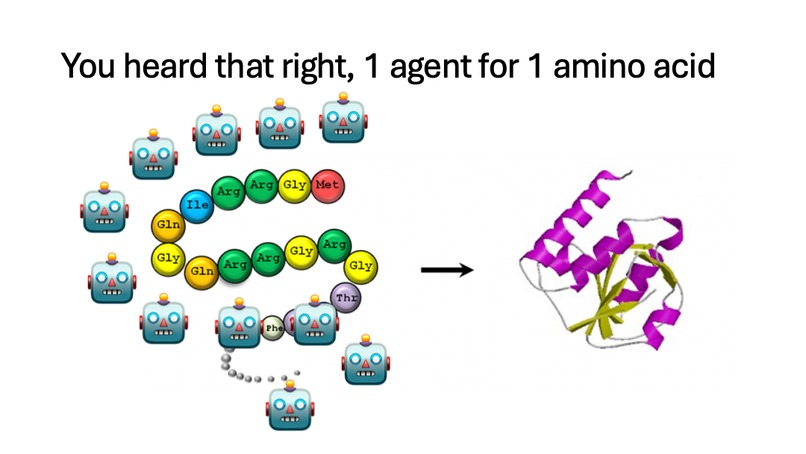
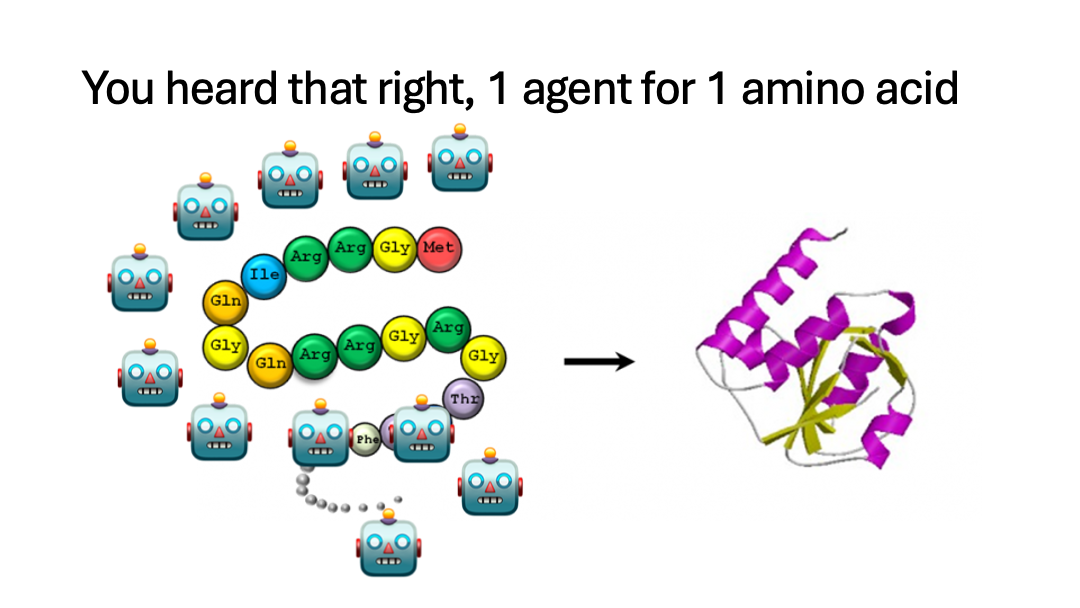
Each Amino Acid in the protein is assigned an agent.
Inspiration
Protein design is still too slow and expensive for most scientists: they iterate between sequence ideas, structure prediction, stability scoring, and “try again” loops, often with limited parallelism. We were inspired by recent research on swarm-based protein design, where many agents work on different residues at once and improve a candidate protein iteratively. BioCol explores that idea with a practical engineering focus: make protein design feel like modern software optimization: fast, iterative, and parallel.
What it does
BioCol (Biological Colony) is a swarm-powered protein design engine that iteratively improves an input amino-acid sequence toward a user-defined objective.
At a high level:
- The user provides an initial protein sequence + a natural-language design goal.
- BioCol spawns one agent per residue position to propose mutations in parallel.
- It merges proposals into a candidate sequence.
- It evaluates candidates using ESMFold for structure prediction and Rosetta (PyRosetta) for stability-aware scoring, with optional structural context from DSSP.
- It accepts or rejects the candidate and repeats until it converges.
How we built it
Core design loop
- Input: amino-acid string + objective prompt.
- Agent swarm: one residue agent per position proposes a mutation + confidence + reason, all running in parallel via Modal's infrastructure
- Merge + constraints: merge mutations into a candidate sequence.
- Evaluation:
- ESMFold on Modal GPU generates a structure report and a confidence score.
- Rosetta computes a physics-inspired energy/stability signal.
- DSSP provides more detailed structure labels.
- We combine physics + objective alignment + confidence into a single score.
- Decision engine: accept if the candidate improves the best score; otherwise reject and learn from memory.
- Memory + throttling: track successes/failures; if repeated rejections happen, the system reduces mutation aggressiveness to avoid unstable jumps and converge faster.
Tech stack
- Python orchestration + CLI
- Modal for distributed agent execution and GPU folding (ESMFold)
- OpenAI API for demo with potential for fully local inference using Modal's infrastructure
- Transformers ESMFold for structure prediction
- PyRosetta for stability scoring
- mkdssp (DSSP) for secondary structure extraction
GUI (Demo) In addition to the backend engine, we built a GUI to make the workflow accessible to researchers:
- Users can enter the initial sequence and natural-language objective.
- Users can configure key parameters (iterations, thresholds, mutation settings).
- The UI visualizes the swarm: agents spawning, getting assigned residue positions, and returning proposed mutations with reasoning.
- It displays the best final protein sequence and renders a 3D view of the predicted structure.
Challenges we ran into
- Convergence: early versions mutated too aggressively and plateaued quickly, so we introduced rejection-aware throttling (dynamic thresholds/mutation rate).
- Learning Curve: learning about and integrating Modal infrastructure for our project was challenging at first, but very rewarding once we figured out the how-to's of it.
- Distributed execution details: running multiple billions-parameter LLM in parallel required careful resource planning and management, effective task execution
Accomplishments that we're proud of
- Built a working multi-agent protein design loop with real parallelism (Modal) and real evaluation (ESMFold + Rosetta).
- Integrated Rosetta scoring to move beyond heuristic-only evaluation.
- Enabled DSSP-based structural context to align with swarm-based protein design prompting.
- Delivered a GUI demo where users can input sequences/objectives, watch agents collaborate in real time, and view the final designed protein (including a 3D structure visualization).
What we learned
- Swarm-style design is less about “many LLM calls” and more about how you coordinate information: memory, evaluation feedback, and controlled exploration matter.
- Protein objectives can conflict (flexibility vs stability), so the system needs clear scoring tradeoffs and guardrails.
- Engineering details (deployment, architectures, binary dependencies) can be just as hard as the ML.
What's next for BioCol
- Improve mutation strategies beyond single substitutions.
- Strengthen the GUI into a full “protein design cockpit” with run history, comparisons across candidates, and clearer structure/energy dashboards.
- Benchmark on known protein design tasks and datasets to validate performance.
Built With
- css
- cursor
- fastapi
- html
- javascript
- modal
- openai
- python
- pytorch
- render
- transformers

Log in or sign up for Devpost to join the conversation.