Inspiration
We wanted our hack to combine some of the domains we are interested in and to solve a small part of an important problem. We've decided to focus on an issue in bioinformatics – handling the data from next generation sequencing.
What it does
We were interested in separation of sequences from biological noise and in data clustering. Traditionally, the methods that are used to solve these problems are heavily parameterized – for example, principal component analysis and hierarchical clustering – making the clustering less robust and requiring precise parameter setup. Our hack consists of a data analysis and visualization package in Python.
The analyzed data were taken from real experiments that look at the mutations occurring during the cycles of evolution of RNA molecules. These are common types of experiments in such fields like virology and origins of life.
How we built it
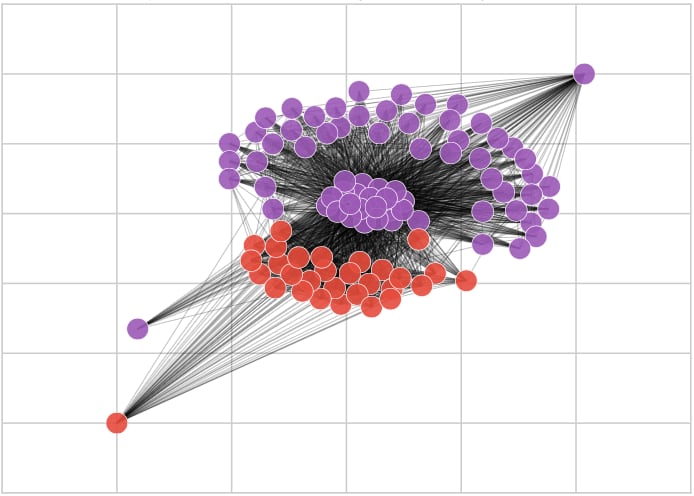
We've developed a method that is based on the graph theory approaches. We represent the sequences as nodes of a graph, where edges and their weight capture how similar these sequences are.

Such representation allows us to use the powerful network science toolbox for further analysis. The initial graph is extremely dense – almost every node is connected to every other. We've removed the edge with insufficient edge weight.

We have used the Louvain Method to discover the underlying community structure of the graph. The method divides the nodes into clusters, maximizing their internal connectivity and minimizing the external connections between them. Such approach is a good fit to our problem, as the related sequences have a high similarity index, translating into high edge weight and, thus, high internal connectivity. The resulting community structure divides the sequences according to their similarities. The method does not require any precise parameter setup and is parallel, making it appropriate for large-scale experiments (we were able to process a graph with 2 million edges on a regular laptop).
We used Python for implementing the method. A number of libraries was used: NumPy, NetworkX, NetworKit, Matplotlib, IPython, and others.
Challenges we ran into
The time required to process large datasets was outside the scope of the event (sequence alignment is very time consuming), thus we had to use medium-sized datasets.
Accomplishments that we proud of
Using experimental RNA data, we were able to demonstrate that such method does achieve its goal – it robustly separates the sequences with the similar mutation profiles. We've tried the method on 7 sets of data. In every case, it yielded satisfactory results.
What we learned
We've gained additional knowledge about bioinformatics and scientific computing.
What's next for BClust
Other network science techniques could be applied to the sequence graphs. We have already considered link prediction and dynamic graph partitioning. We are hoping to validate our technique on a larger dataset.

Log in or sign up for Devpost to join the conversation.