-
-
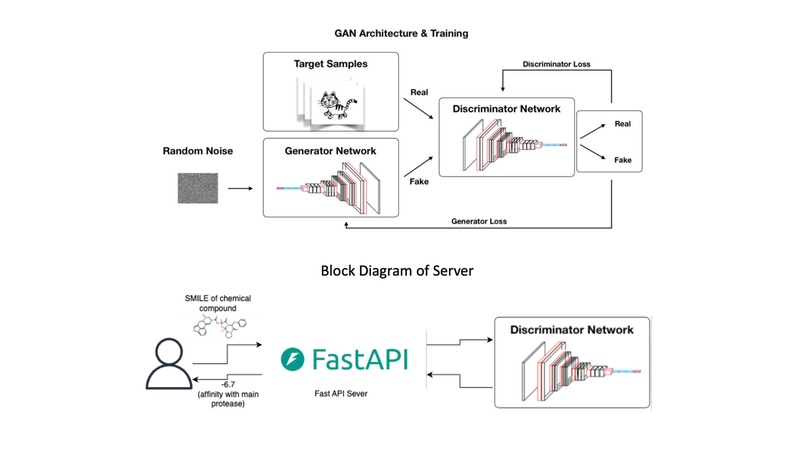
Block Diagram
-
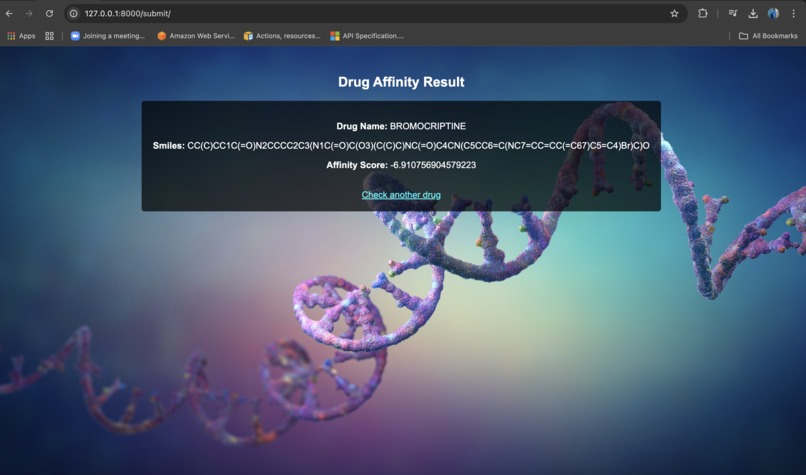
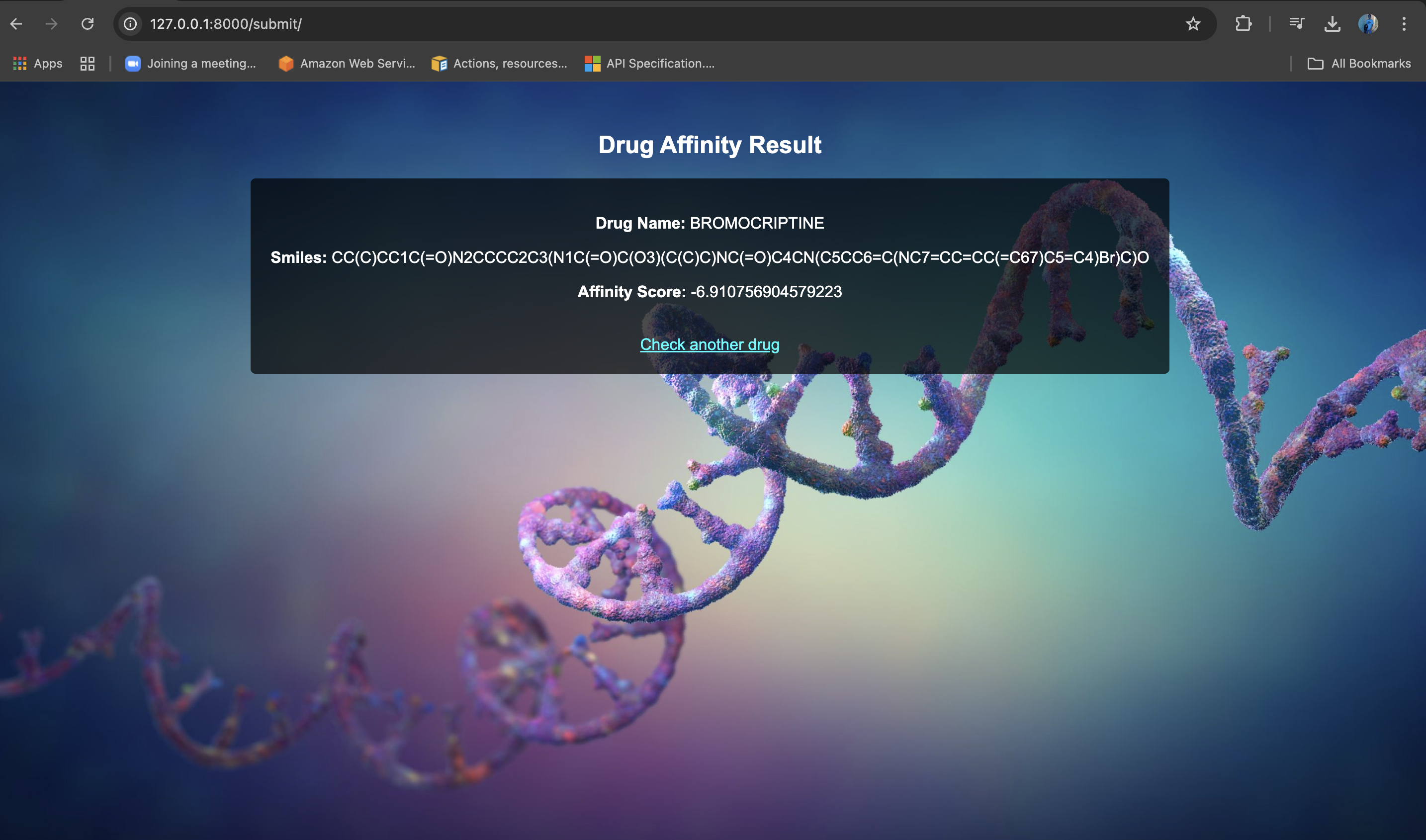
Web UI

🧪 AI-Driven Drug Repurposing & Molecular Generation
🌟 Inspiration
The COVID-19 pandemic exposed the urgent need for faster drug discovery methods. With no specific cure and new variants emerging rapidly, traditional drug development—which takes 5-10 years and millions of dollars—was not feasible.
We were inspired by:
- The success of drug repurposing in past pandemics.
- The advancements in AI & Machine Learning (ML) for computational drug discovery.
- The potential of Generative Adversarial Networks (GANs) to design new drugs with higher efficacy.
This project was born from a desire to accelerate drug discovery using AI while keeping costs low and making treatments more accessible.
🔬 What It Does
Our solution integrates machine learning and AI-driven molecular generation to:
- Identify repurposed drugs that can inhibit the SARS-CoV-2 main protease.
- Predict the binding affinity of any given drug molecule using a Random Forest Regressor.
- Generate new molecular compounds with high binding affinity using a Generative Adversarial Network (GAN).
- Provide a user-friendly web application where researchers can:
- Input a drug name or chemical formula
- Get a predicted binding affinity score
- Discover alternative drug candidates
- Input a drug name or chemical formula
🏗 How We Built It
1️⃣ Data Collection & Processing
- Extracted data from ChEMBL and docking simulations.
- Filtered and preprocessed 9001 molecules with valid SMILES representations.
2️⃣ Binding Affinity Predictor (Discriminator Model)
- Used mol2vec embeddings to convert molecules into numerical fingerprints.
- Developed a Random Forest Regressor trained on AutoDock Vina scores.
- Evaluated using Mean Absolute Error (MAE), Mean Squared Error (MSE), and R² score.
3️⃣ Drug Design Using Generative Adversarial Network (GAN)
- Designed a Stack-Augmented Recurrent Neural Network (RNN) to generate molecular structures.
- Used GRU (Gated Recurrent Units) to handle sequential SMILES data.
- Set hidden layers = 1500, depth = 200 layers, and learning rate = 0.01.
- The discriminator (Random Forest Regressor) assessed their binding affinity.
- Generated 10,000 molecules, out of which 6,321 were valid.
4️⃣ Web Application
- Deployed a Flask/Django backend with a React frontend.
- Allows users to search for a drug by name or chemical formula and retrieve predictions.
🚧 Challenges We Ran Into
Handling Large Molecular Data
- Processing thousands of molecules and converting them into meaningful vectors was computationally intensive.
- Solution: Used high-performance computing (HPC) and optimized memory management.
- Processing thousands of molecules and converting them into meaningful vectors was computationally intensive.
Optimizing the GAN Model
- Initially, the model generated invalid molecular structures.
- Solution: Fine-tuned the RNN layers, stack depth, and learning rate to improve molecular validity.
- Initially, the model generated invalid molecular structures.
Ensuring Model Generalization
- The Random Forest Regressor needed to generalize well on unseen molecules.
- Solution: Used cross-validation, hyperparameter tuning, and feature selection.
- The Random Forest Regressor needed to generalize well on unseen molecules.
Deploying the Web Application
- Making the model accessible via a simple web interface required integrating ML predictions into a user-friendly UI.
- Solution: Used Flask/Django for API and React for frontend.
- Making the model accessible via a simple web interface required integrating ML predictions into a user-friendly UI.
🏆 Accomplishments That We're Proud Of
✅ Successfully trained a Random Forest Regressor to predict drug binding affinity.
✅ Developed a GAN model that can generate new drug-like molecules.
✅ Built a fully functional web application to make AI-driven drug discovery accessible.
✅ Generated 6,321 valid molecular compounds, some with higher predicted binding affinity than existing drugs.
✅ Optimized AI algorithms to work efficiently on large molecular datasets.
📚 What We Learned
🔬 Drug Discovery: How AI can accelerate molecular screening & drug repurposing.
💡 Molecular Fingerprinting: Using mol2vec embeddings for molecular structure representation.
🧠 Deep Learning: Training a GAN model for molecular generation.
⚡ High-Performance Computing (HPC): Handling large-scale molecular datasets.
🌐 Web Deployment: Integrating ML models into a real-world application.
🚀 What's Next for AV_Algos
🔹 Laboratory Testing: Synthesizing and testing AI-generated molecules for real-world efficacy.
🔹 Integration with High-Throughput Screening: Combining AI models with wet lab experiments to accelerate drug discovery.
🔹 Improved Molecular Generation: Exploring transformers instead of RNNs for better SMILES sequence generation.
🔹 Expansion to Other Diseases: Applying the same methodology to cancer, HIV, and other viral infections.
🔹 Enhancing the Web Application: Adding features like drug similarity searches and real-time docking simulations.
📌 Key Takeaway:
💡 AI-driven drug repurposing and molecular generation can significantly accelerate the discovery of new treatments for emerging diseases like COVID-19.
🌍 Our web app makes this technology accessible to researchers worldwide.
Built With
- colab
- fastapi
- python

Log in or sign up for Devpost to join the conversation.