
Inspiration
Unfortunately, none of us on the team and our families are strangers to diseases such as cancer. With this tool, we aim to accelerate biomedical research by accurately identifying drug candidates, increasing the accuracy of medical therapies and driving collaboration in science.
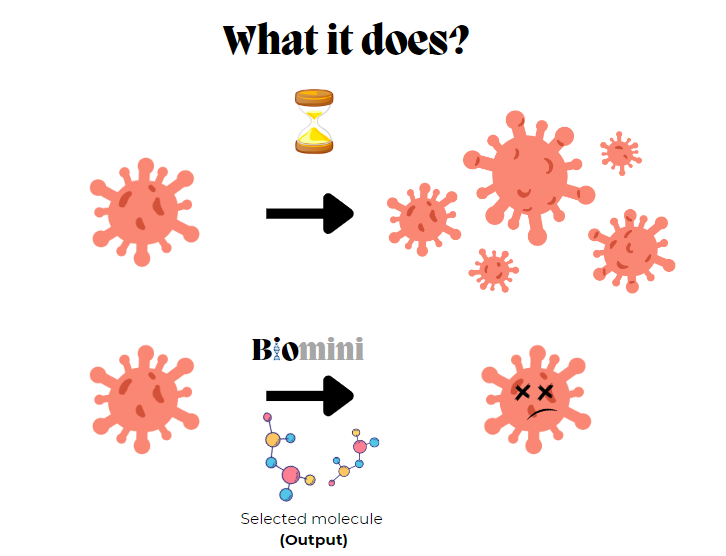
What it does
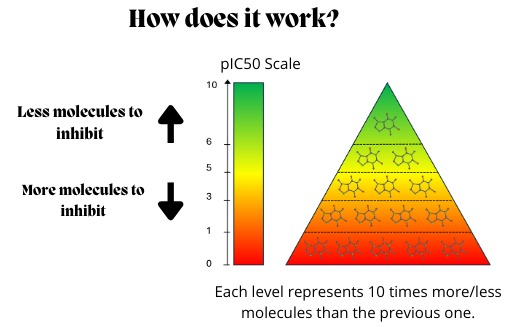
We have developed a web platform that: 1) Automates the construction of machine learning models for predicting the pharmacological potency of drugs (pIC50) 2) Evaluates molecules approved by organizations worldwide and suggests those with potential for drug repurposing. 3) Mutates the most bioactive molecules identified to find more specific compounds with fewer side effects. 4) Allows the user to test their own molecules in the trained models.
How we built it
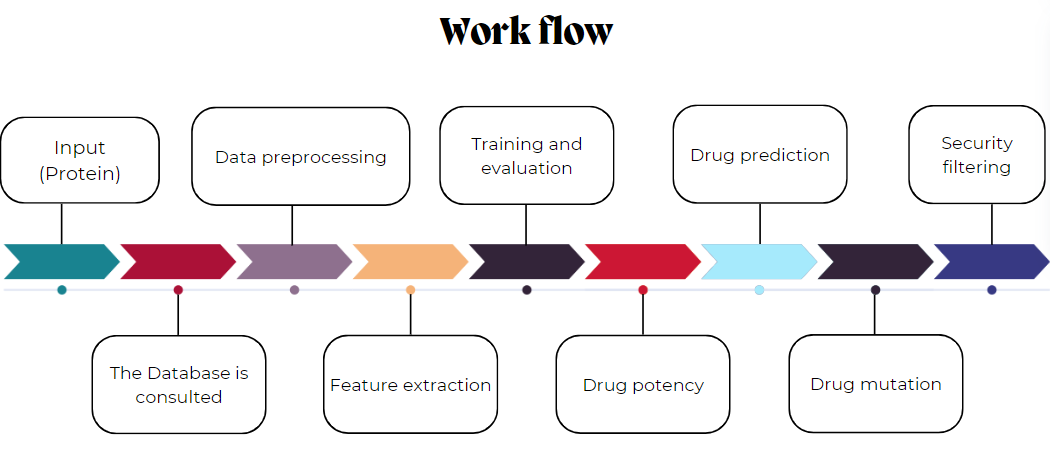
The process starts with the query of the target protein data (virus, bacteria, etc). Then, a specific protein is selected and its behaving data is downloaded. Data is cleaned by removing duplicates and missing values. Next, important features are extracted, values are processed to enrich the data. Descriptive statistics are displayed and several regression models are trained to choose the best one. The strongest molecules are identified and modified to evaluate whether they can be improved. Finally, it lets the user to test their own molecules by loading a csv and specifying the data.
Challenges we ran into
Molecular design is a complex topic that makes it difficult to simplify, communicate with professionals not dedicated to this area. It also includes learning front-end design with Streamlit, developing a functional backend, automating a pipeline, and creating multimedia content to explain the results obtained after running simulations.
Accomplishments that we're proud of
We materialized an idea that at the beginning of the day seemed impossible, with teamwork, communication and effort we achieved to see encouraging results in a short period of time. We hope that in the future, our work will be useful for the scientific community on the development of new treatments, reducing the design time of new drugs and increasing the number of patients treated.
What we learned
We learned how to efficiently query and select target data, manage and preprocess the potency data, and calculate crucial chemical features. By preparing the data accordingly, we were able to train and evaluate several regression models, finally selecting the best one based on performance metrics. This process allowed us to predict values for new data of molecules, demonstrating the application of machine learning in bioactivity predictions and improving our ability to interpret and visualize molecular data. Also the ability of encoding/decoding models to generate valid mutants.
What's next for Biomini
Development of the tool in cloud services so that the scientific community can use it, in this way we would obtain feedback from them for the future optimization of our project. For the statistical models within the tool, deep learning could be implemented to increase their accuracy, and they could also be fed with a larger amount of data, handling more options.
Built With
- chembl
- css
- database
- google-colab
- linux
- python
- rdkit
- sklearn
- streamlit
- visual-studio


Log in or sign up for Devpost to join the conversation.