-
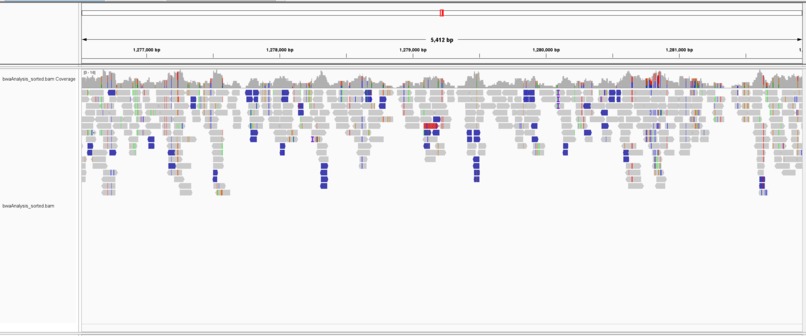
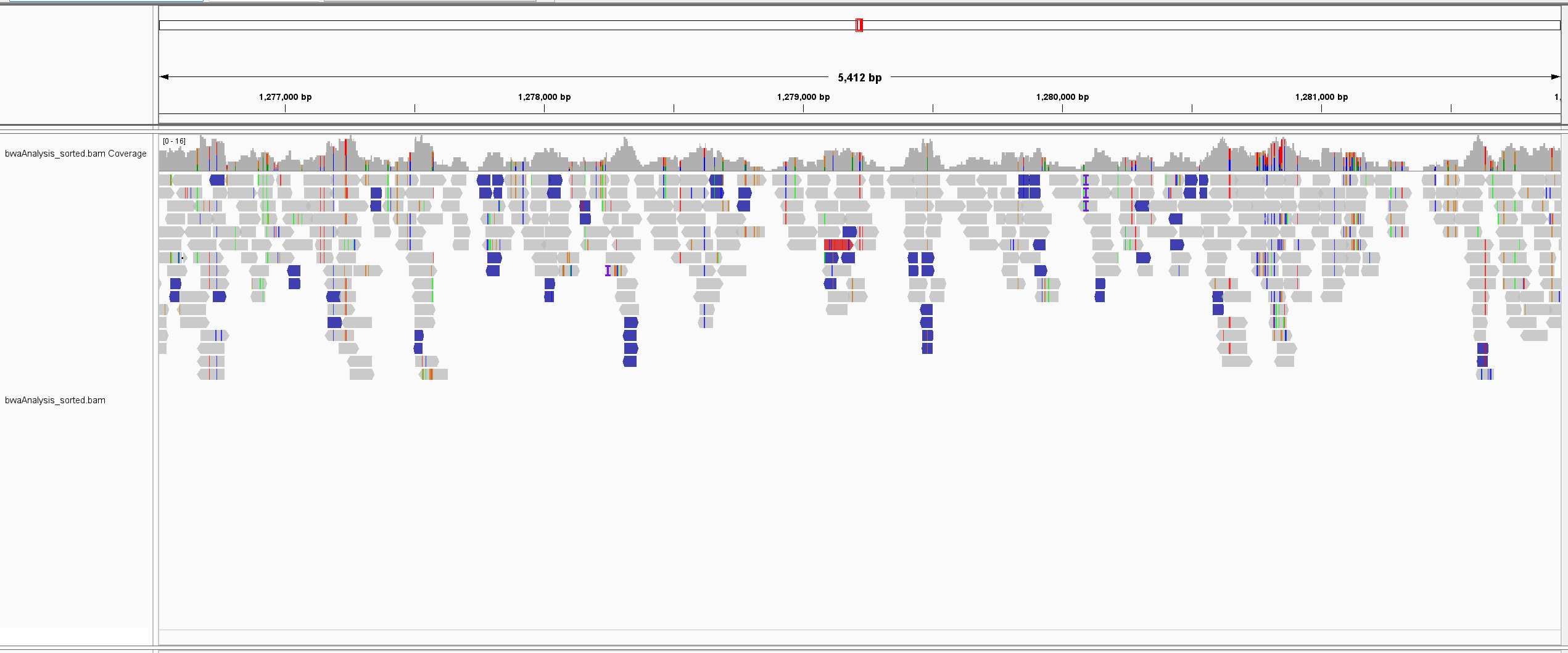
IGV Visualization of BWA
Inspiration
Computational Biology allows for the intersection of biology and computing for technological innovations and optimizations in genomics, modeling systems, biology, phylogenetics, etc. In our project, we chose to focus on studying the structure and function of sequences from a community of organisms, like on human skin, in the soil, or in a water sample aka metagenomics. Computational analysis in metagenomics is based on sequencing data, samples are taken from microbiomes. These samples are then sequenced to find out what the nucleotide(DNA) or amino acid (protein) sequences are. This is the information which is analyzed and draws conclusions from using computational tools. Sequencing technologies have given us the ability to gather sequence data relatively efficiently and cheaply. One important task we need to do is compare sequences to each other - not all organisms will have the same exact sequences with the same letters in the same position. To be able to compare these sequences, we use alignment tools! Alignment tools help us find regions of similar or identical sequences between DNA/RNA/protein sequences. Pairwise sequence alignments compare two sequences at a time to find overlap. However, most of the time, we need to examine the sequences of many organisms at a time so we use MULTIPLE SEQUENCE ALIGNMENT (MSA) tools!
What it does
Our research analyzed the alignment rates and how long it takes for each tool to compare the data between the reference files and our reads.
How we built it
We learned how to use various applications including Bowtie2, BBMap, and BWA to analyze our genome samples in comparison to our reference file. Through these applications, we were able to collect data that showed us the alignment rates and how long it took for each tool to analyze the data on each of our computers. We converted the output files and inputted the reference files and output files into the IGV visualization tool to see a visual representation of the alignments.
Challenges we ran into
The main challenges we faced were properly downloading and making sure each tool and Anaconda would run properly onto our laptops and ensuring that our scripts were running properly. Since each team member had a different machines, we had some trouble with finding commands which would work on each device and also each device took a different amount of time to process the mapping. Ultimately, we all worked together to fix these issues and were able to get our applications and tools working to complete our project.
Accomplishments that we're proud of
We were able to successfully run all three tools on different machines and found interesting results. We were also able to understand and reapply algorithms used throughout the different tools.
What we learned
We learned how to create an anaconda environment to be able access all alignment tools, how common alignment tools are used in computational biology, how to look at tutorials/githubs to find out how to run these alignment tools, and how to measure time and memory it takes to run each alignment tool.
What's next for The Fundamentals of Metagenomics
We will continue to explore different computational tools and engage in the bioinformatics research field. Using resources from our mentors and online we will continue to pursue this passion.






Log in or sign up for Devpost to join the conversation.