-
-
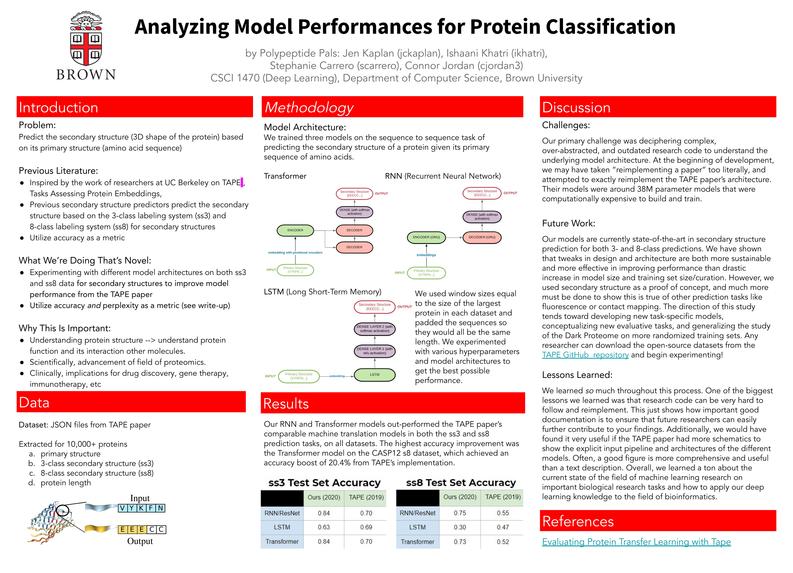
Analyzing Model Performances for Protein Classification Poster
Analyzing Model Performances for Protein Classification Final Write-Up
Hi everyone! We are the Polypeptide Pals and we will be tackling the quandary of protein modeling by harnessing the learning capacity of transformers and other neural networks. Being able to predict the structure, connectivity, and other properties of proteins through sheer computational guesswork is near impossible. However, understanding these properties can result in critical contributions to drug development and even the future of personalized medicine. Thus, we have decided to re-implement a study that examines the performance of three types of neural networks in predicting properties such as secondary structure, evolutionary relationships, stability, and more based on the amino acid sequence of a protein.
Group Members: Jen Kaplan (jckaplan) Hi! I’m Jen and I’m a Software Engineer and part-time 5th Year CS Master’s student studying AI/ML. I have no prior experience with computational biology, but am very interested in the application of deep learning to bioinformatics.
Connor Jordan (cjordan3)
Ishaani Khatri (ikhatri) Hello! I’m Ishaani and I am a 4th year undergraduate concentrating in computational biology. I’ve done a few computational biology projects before, but never with protein data or utilizing deep learning, so I’m very excited for this project!
Stephanie Carrero (scarrero) Hi! I’m Stephanie and I am a 4th-year studying computer science and biology. I’m interested in developing technological solutions to biomedical and clinical challenges.
Introduction:
Hello Deep Learners! We are the Polypeptide Pals. For our final project, we will be reimplementing the paper Evaluating Protein Transfer Learning with TAPE. Protein modeling is an emerging field in deep learning research with enormous potential. The size of protein datasets has been exponentially increasing in recent years due to the advancement of sequencing technologies. However, there is a growing gap between the size of those datasets and their labeled subsets. The reason for this is that it takes massive amounts of time, experimental equipment and scientific expertise to label and annotate these proteins in meaningful ways. With NLP models, we can automate the extraction of this useful biological information from sequence datasets, leading to potential rapid advances in structural bioinformatics and genomics. Such advances include structure prediction, detection of remote homologs, and protein engineering.
We chose this paper because we were all interested in the applications of deep learning to solve biological problems. Specifically, some group members had the experience and biological background working with proteomic data before. As a result, we thought it would be good to focus on a project related to proteins in order to leverage our group members’ knowledge in the field.
This paper is a self-supervised classification problem that assesses the performance of three models (LSTM, ResNet, and a Transformer) on five biologically relevant semi-supervised learning tasks spread across different domains of protein biology. The objective of the paper is to determine their potential on correctly predicting the biological features of these input sequences. The paper has a GitHub repository holding access to their dataset and model architecture built using PyTorch. We will be reimplementing one of their models in Tensorflow.
The tasks presented in the paper are described below:
Task 1: Secondary Structure (SS) Prediction (Structure Prediction Task) “SS is an important feature for understanding the function of a protein, especially if the protein of interest is not evolutionarily related to proteins with known structure Task 2: Contact Prediction (Structure Prediction Task)” “Accurate contact maps provide powerful global information; e.g., they facilitate robust modeling of full 3D protein structure” Task 3: Remote Homology Detection (Evolutionary Understanding Task) “Detection of remote homologs is of great interest in microbiology and medicine; e.g., for detection of emerging antibiotic resistant genes and discovery of new CAS enzymes” Task 4: Fluorescence Landscape Prediction (Protein Engineering Task) “For a protein of length L, the number of possible sequences m mutations away is O(L m), a prohibitively large space for exhaustive search via experiment, even if m is modest. Moreover, due to epistasis (second- and higher-order interactions between mutations at different positions), greedy optimization approaches are unlikely to succeed. Accurate computational predictions could allow significantly more efficient exploration of the landscape, resulting in better optima.” Task 5: Stability Landscape Prediction (Protein Engineering Task) “Designing stable proteins is important to ensure, for example, that drugs are delivered before they are degraded. More generally, given a broad sample of protein measurements, finding better refinements of top candidates is useful for maximizing yield from expensive protein engineering experiments.”
Related Work
Our project seeks to address the challenge of predicting 3-D structure from a protein’s primary sequence in order to aid in the drug development process. Many other researchers aim to take the opposite approach to drug engineering, which is to concoct a 3-D structure and apply computational models to determine the primary sequence that can be used to engineer their prototype molecule. In this paper entitled “Generative models for graph-based protein design” researchers represented 3-D protein structures as graphs and devised a deep conditional generative model to reduce the graph to its primary amino acid sequence. Research studies such as this one represent one of the key goals of computational protein design, which is to automate protein engineering based on desired protein structure and function.
Additionally, linked here is the public GitHub for the paper we are trying to implement: https://github.com/songlab-cal/tape#list-of-models-and-tasks
Data:
The data we will be using in our reimplementation is the data provided on the paper’s GitHub. This data is publicly available and accessed via a download script. The sequence data is 120MB compressed and 900MB uncompressed. In terms of preprocessing, much of the preprocessing done in the paper involves splitting the dataset into training, validation, and test sets at varying ratios based on the dataset and task. For datasets being used to examine remote homology, or the evolutionary relationships between proteins, proteins are classified in superfamilies and entire superfamilies were taken out of the training dataset to test the model’s capacity for generalizing these relationships. Overall, the main preprocessing step discussed in the article is data splitting.
Methodology:
The paper implements three different model architectures, however, due to time and resource constraints, we will only be implementing one, specifically a transformer model. We will train the model using Google Cloud Platform on four NVIDIA V100 GPUs (to mimic the paper’s hardware). The paper states that each of their model implementations took one week to train. Since we only have four weeks to complete our reimplementation and since all of their models are extremely robust in terms of the size of the hyperparameter space, we will attempt to replicate one of their models, and may reduce the size of the model to decrease the train time.
Their Transformer model implements a 12-layer architecture with a hidden size of 512 and 8 attention heads. This model has 38 million parameters. Our primary challenge here is managing the size of the dataset and sheer number of parameters of this model within such a short time period. Rao et al’s LSTM model consists of two three-layer LSTMs with a hidden size of 1024 and follows a similar architecture as ELMo. Finally, their ResNet implementation used 35 residual blocks, each containing two convolutional layers with 256 filters, a kernel size of 9 and a dilation rate of 2.
If we run into issues with building such a complex and computationally expensive model, we may reduce the size of the hyperparameter space of our implementation. While this may reduce the accuracy of our results, we will be satisfied with getting a working model that proves the efficacy of NLP models on predicting protein structures. We will also try to experiment with different optimizations to the paper’s model architectures that do not include the size of the hyperparameter space.
Metrics:
We plan to focus on a single task from the five tasks presented in the paper to reduce train time and to attempt to replicate their results. Our target goal is to implement one of their model’s architectures and get equal or similar results to the paper on a single task. While this may not be doable in the allotted time, we see success in this final project as simply implementing one of their models and getting results that prove the efficacy of the model on the task we focus on.
Since this is a supervised classification problem, accuracy and perplexity do apply for our project, and we hope to get decent results. However, we understand that because of the size of the model needed to achieve those results, we may not be able to reach the same level of accuracy that is presented in the paper’s results. Accuracy was calculated for each model and task differently, with some metrics being determined on different protein families, lengths, or other categories. The best performing model in the paper was their Transformer architecture (with pre-training), which achieved an overall rank correlation coefficient (also known as Spearman’s p) of 68% and 73% respectively for the fluorescence and stability tasks.
Our base goal is to reimplement one of the architectures from the paper, specifically a smaller transformer model, and achieve comparable results on a single task given our time/resource constraints. Our target goal is to reimplement one of the architectures and replicate the results on a single task. Our stretch goal is to reimplement one of the architectures and replicate the results on all five biological tasks.
Ethics:
Proteins are essential to the function of our cells. For proteins, like many other molecules, form follows function. Thus, protein malfunction underlies many diseases. Understanding protein structure is vital to understanding disease. By building a protein structure predictor, we will be able to understand how protein structure is maintained and built. This predictor has the potential to understand how the change in protein sequences affects protein structure, which is a key biological question.
Additionally, the protein engineering tasks are important because they have implications for drug delivery and pharmaceutical development. If we understand how likely proteins are to degrade and maintain their fluorescence or stability, we will be able to design more effective and efficient pharmaceuticals.
Thus, the pharmaceutical industry is a large stakeholder for this project. If our algorithm has mistakes, the pharmaceutical industry could spend lots of money developing ineffective, or even unsafe, drugs. For instance, our algorithm could suggest a certain molecular compound would be beneficial to treat a certain disease, when it in reality is not. Thus, the company would spend lots of money investigating a pharmaceutical treatment that may not be that stable or that safe.
Division of labor:
To manage the project workload, we have made a Trello board with tasks and plan to implement an Agile development process, where group members will pick up tasks freely. We have not yet concretely divided up the work, as we plan to have each group member participate in each step of the process. Also, since we have not yet begun coding, we do not know the extent of each step of the model implementation. As this becomes more clear, we may assign group members to larger aspects of the project.
Built With
- casp
- numpy
- tensorflow


Log in or sign up for Devpost to join the conversation.