-
-
poster
Deep learning for single-cell age prediction
Our final report is at: https://docs.google.com/document/d/1cPdudc_rjnOkndqY5SGkCPj2Dla8iWH6bRxHrXG4rpU/edit
Introduction:
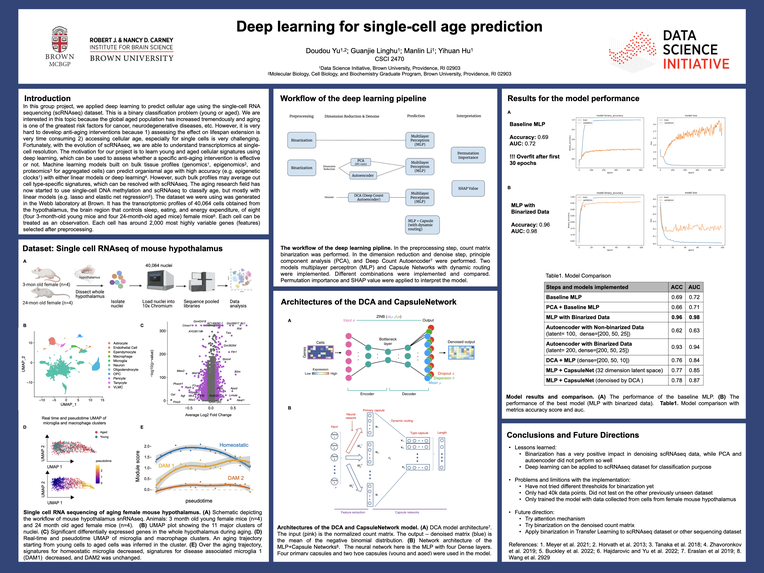
In this group project, we are trying to apply deep learning to predict cellular age using the single-cell RNA sequencing (scRNAseq) dataset. This is a binary classification problem (young or aged). We are interested in this topic because the global aged population has increased tremendously and aging is one of the greatest risk factors for cancer, neurodegenerative diseases, etc. However, it is very hard to develop anti-aging interventions, partially due to the difficulties in accessing cellular age, especially for single cells. With the evolution of scRNAseq, we are able to understand transcriptomics at single-cell resolution. The motivation for our project is to learn young and aged cellular signatures using deep learning, which can be used to assess whether a specific anti-aging intervention is effective or not.
Related Work:
Machine learning models built on bulk tissue profiles (genomics[1], epigenomics[2], and proteomics[3] for aggregated cells) can predict organismal age with high accuracy (e.g. epigenetic clocks[1]) with either linear models or deep learning[4]. However, such bulk profiles may average out cell type specific signatures, which can be resolved with scRNAseq. The aging research field has now started to use single-cell DNA methylation and scRNAseq to classify age, but mostly with linear models (e.g. lasso and elastic net regression[5]). The previous work mentioned above can help our project in terms of how to perform data preprocessing and how to avoid potential pitfalls in age prediction.
Data:
The dataset we are using was generated in the Webb laboratory at Brown. It has the transcriptomic profiles of 40,064 cells obtained from the hypothalamus, the brain region that controls sleep, eating, and energy expenditure, of eight (four 3-month-old young mice and four 24-month-old aged mice) female mice[6]. Each cell can be treated as an observation. Each cell has around 3,000 highly variable genes (features) selected after preprocessing.
Methodology:
Currently, in the field of single-cell DNA methylation and scRNAseq, linear models such as lasso and elastic net regression are applied. The aim of this project is to explore how powerful non-linear models can be utilized in aging prediction with different deep learning techniques. This aging prediction model begins with a simple multi-layer perceptron to learn the hidden nonlinear pattern. We would like to explore the potential power of CNN, RNN, and transformers in the latter analysis. Even though those models cannot be interpreted explicitly, we believe that the accuracy score would be higher than current models. In case we ran into the issue related to the noise from scRNAseq amplification and dropout, one backup plan is to apply the autoencoder to denoise our data [7].
We start with Exploratory Data Analysis to have a basic idea of the data. The next step is data preprocessing including handling missing data and normalization. The data is split for 60-20-20: 60% of data used to train the model, 20% for validation to tune parameters, and 20% for test data. Since the database only has around 40k cells, cross-validation is necessary to compensate for the small size of the original dataset and avoid the variability of random splits. The number of epochs should be decided during the training process. After splitting, we would take PCA or TSNE to reduce dimension among 3000 features. Then, models are implemented using MLP, CNN, RNN, and transformers. For each model, hyperparameters are chosen using validation scores. Lastly, the best model is decided based on the test score.
Metrics:
The metric used to evaluate the model in the hyperparameter tuning process and model comparing process is accuracy since we are trying to solve a binary classification problem and our data set can be considered as balanced, with a class ratio of about 6:4. Also, as per the zero rule, the accuracy of predicting the majority class can be used as our baseline model. AUROC is another evaluation metric that we use to complement the accuracy. The unbiasedness of AUC can help us with the little imbalance of the data set.
Base: Get a better performance than the baseline model.
Target: Outperform linear models in previous works.
Stretch: Generalize our model to different data sets and different types of cells.
Ethics:
(1) What broader societal issues are relevant to your chosen problem space?
Although the main focus of our project is to predict the aging of single-cell of mice, the broader application of it will be predicting that of human beings. In that case, discrimination may exist in the health care industry if institutions have easy access to people’s chronological and biological age. For example, research suggests that for every 5 years a woman’s biological age is older than her chronological age, her risk of breast cancer increases by 15%[8]. If the access to patients’ personal information on their chronological and biological age is without limitation, they may be treated differently such as receiving different quotes from insurance companies.
(2) What is your dataset? Are there any concerns about how it was collected, or labeled? Is it representative? What kind of underlying historical or societal biases might it contain?
Our dataset is collected from mice, and the scRNAseq dataset was generated using the droplet-based scRNAseq technology from 10x. Again, when we generalize the application of this project to broader areas including human aging, the collecting process should follow legitimate standards and individual consent. Additionally, racial discrimination may also exist due to the samples collected from different races, which is similar to the one that existed in facial recognition models. Therefore, more attention should be paid to this underlying societal bias.
Division of labor:
Data preprocessing: Doudou
Data splitting and dimension reduction: Guanjie
MLP: Doudou
CNN: Guanjie
RNN: Yihuan
Transformers: Manlin
Citations
Meyer, David H., and Björn Schumacher. "BiT age: A transcriptome‐based aging clock near the theoretical limit of accuracy." Aging cell 20.3 (2021): e13320.
Horvath, Steve. "DNA methylation age of human tissues and cell types." Genome biology 14.10 (2013): 1-20.
Tanaka, Toshiko, et al. "Plasma proteomic signature of age in healthy humans." Aging cell 17.5 (2018): e12799.
Zhavoronkov, Alex, et al. "Deep biomarkers of aging and longevity: from research to applications." Aging (Albany NY) 11.22 (2019): 10771.
Buckley, Matthew T., et al. "Cell type-specific aging clocks to quantify aging and rejuvenation in regenerative regions of the brain." bioRxiv (2022).
Hajdarovic, Kaitlyn H., et al. "Single cell analysis of the aging hypothalamus." bioRxiv (2021).
Eraslan, Gökcen, et al. "Single-cell RNA-seq denoising using a deep count autoencoder." Nature communications 10.1 (2019): 1-14.
Older Biological Age Compared to Chronological Age Linked to Increased Breast Cancer Risk.
Reflection:
Introduction: In this group project, we are trying to apply deep learning to predict cellular age using the single-cell RNA sequencing (scRNAseq) dataset. This is a binary classification problem (young or aged). We are interested in this topic because the global aged population has increased tremendously and aging is one of the greatest risk factors for cancer, neurodegenerative diseases, etc. However, it is very hard to develop anti-aging interventions, partially due to the difficulties in accessing cellular age, especially for single cells. With the evolution of scRNAseq, we are able to understand transcriptomics at single-cell resolution. The motivation for our project is to learn young and aged cellular signatures using deep learning, which can be used to assess whether a specific anti-aging intervention is effective or not. Challenges: The hardest part of our project is choosing the appropriate model. We were thinking about CNN and transformers. Sadly, the top 2000 features (the most highly variable genes) we selected did not have local structures or order. Unlike images or sentences which have local features for models to learn, genomic dataset seemed to have features unrelated to each other at the first glance. Given that challenge, we first tried some simple multi-layer perceptrons (MLP).
Insights: Are there any concrete results you can show at this point? How is your model performing compared with expectations? Machine Learning Part: We implemented XGBoost for baseline comparison. Deep Learning Part: So far, we have implemented 2 versions of MLP: (1) MLP (2) MLP with PCA. For the first one, there’s clear pattern of overfitting in which the training accuracy reaches 0.99 while the testing accuracy is stuck at ~0.6 after about 30 epochs. For the second one, although the test accuracy is still not hight (around 0.6), but we can see clear pattern of convergence.
Plan: Are you on track with your project? What do you need to dedicate more time to? What are you thinking of changing, if anything? Since the current model did not achieve high performance, more time should be dedicated to tune hyperparameters such as learning rate and number of layers for MLP. Besides, autoencoders should be implemented in the dimension reduction process and should be compared with the implemented PCA. If all of those tricks don’t work, we’re also thinking of manually selecting some features before fitting the model.
Built With
- python
- tensor




Log in or sign up for Devpost to join the conversation.