-
-
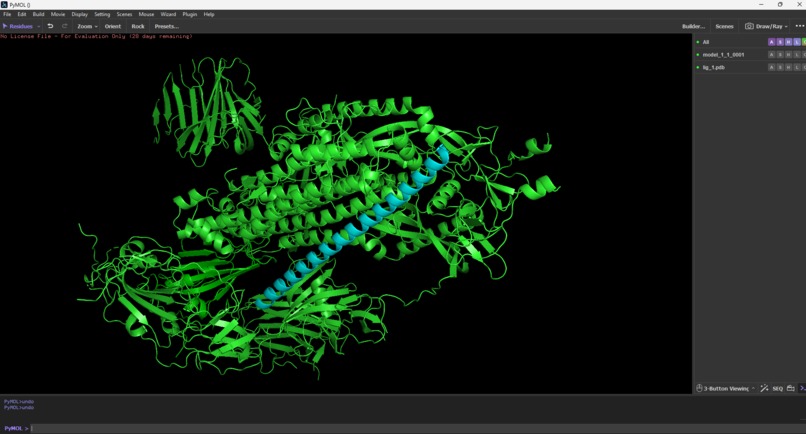
AEGIS_V007 vs SARS-CoV-2 Spike Protein
-
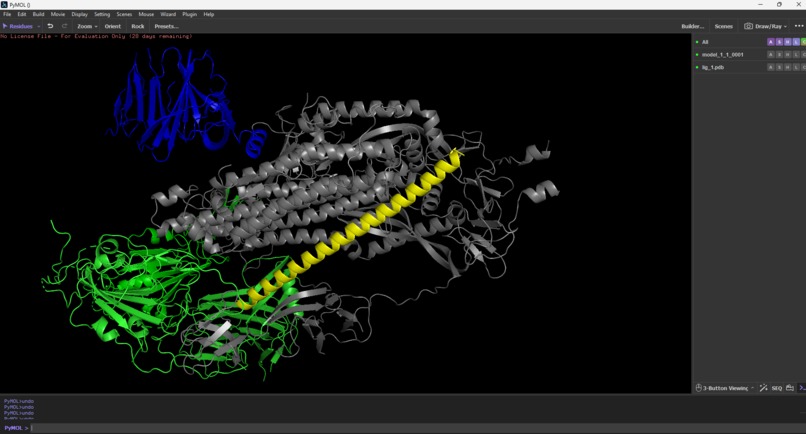
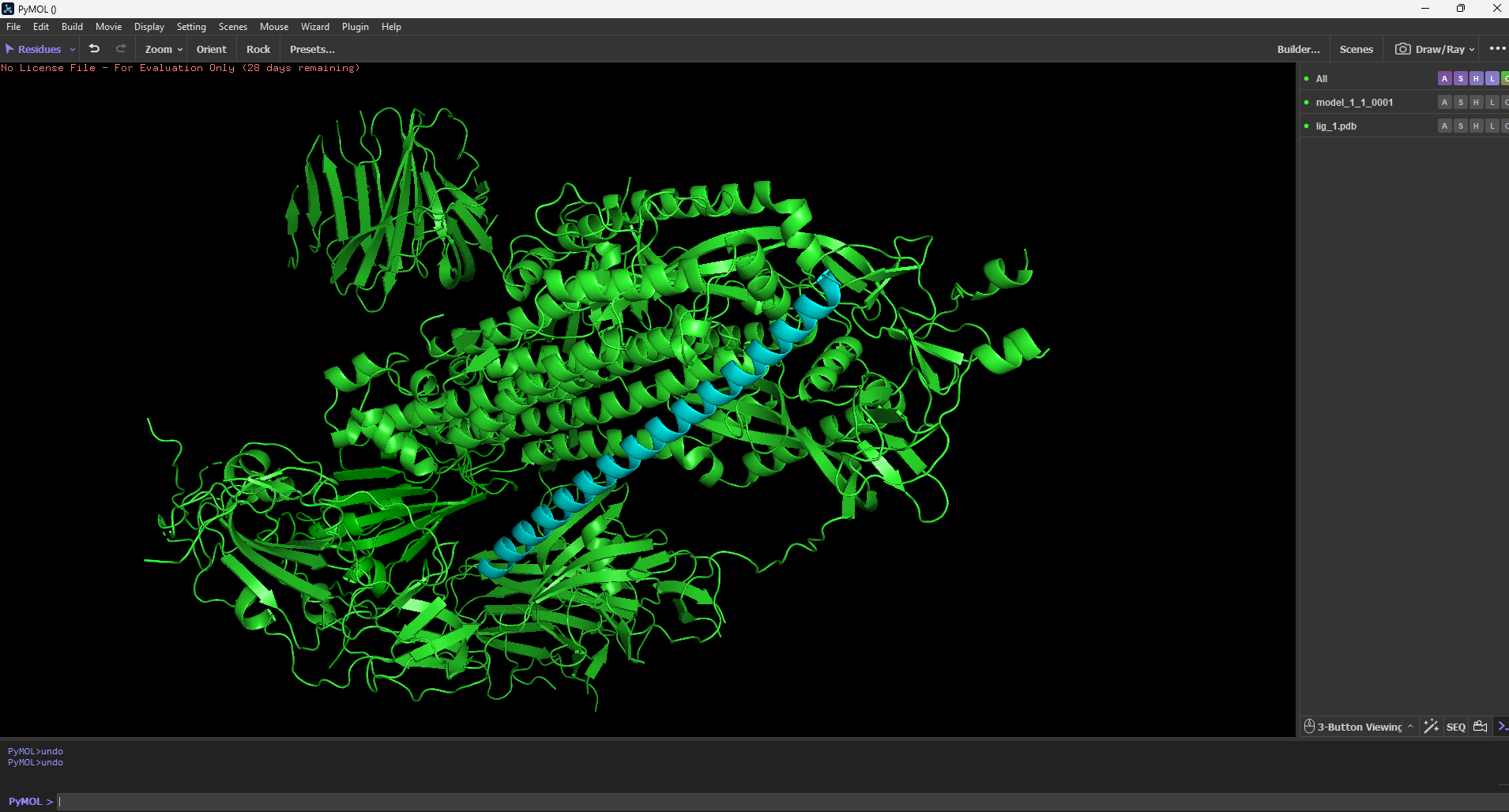
PyMol software used for testing AEGIS_V007 vs SARS-CoV-2 Spike Protein
-
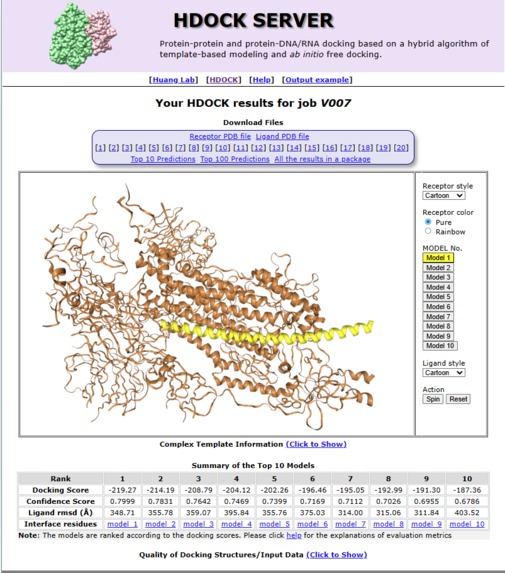
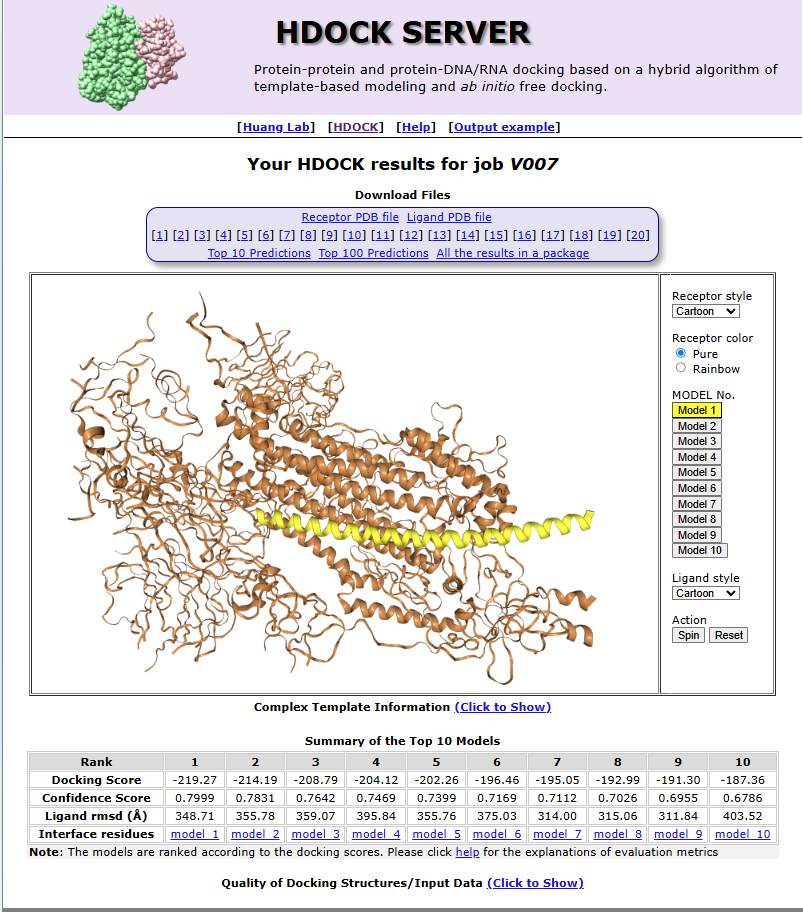
HDOCk analysis
-
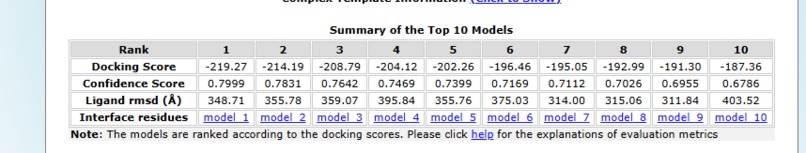
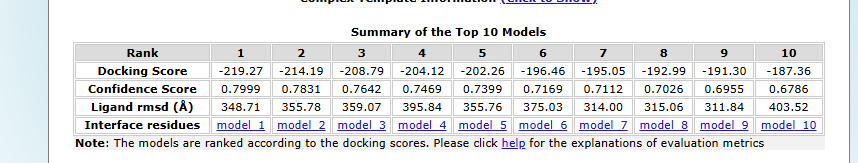
Docking analysis table
Inspiration
The idea for AEGIS was born when I thought about the chaos of the COVID-19 pandemic. I watched as the world came to a standstill, waiting months for a vaccine while the virus spread unchecked.
Although the SARS-CoV-2 genome was sequenced within weeks, the earliest stages of therapeutic discovery—target analysis, candidate design, and prioritization—still took months. That delay exposed a critical weakness in our global response: human-driven early discovery does not scale to pandemic speed.
I asked a simple question: Why does the earliest phase of drug discovery still depend on manual reasoning when we now have powerful generative and structural AI models?
AEGIS is my answer. It is a "digital immune system" — a system that activates immediately after a pathogen is sequenced and begins generating credible, structurally validated therapeutic hypotheses within minutes, not months.
AEGIS does not replace biology or clinical trials. It accelerates the slowest part of the pipeline so biology can begin sooner.
What it does
AEGIS is an autonomous computational discovery platform that compresses the earliest phase of therapeutic design—from months to minutes—by generating, filtering, structurally predicting, and docking candidate protein binders against viral targets.
Input: A raw genomic FASTA file (e.g., a newly sequenced pathogen genome) Output: Structurally validated protein binder blueprints with predicted 3D structures and molecular docking scores, ready for experimental synthesis and testing.
AEGIS focuses exclusively on the preclinical ideation and validation phase, where time savings have the highest leverage.
The Watchtower (Pathogen Analyzer):
- Ingests a raw genomic FASTA file
- Uses AI to identify the virus and its primary therapeutic target protein
- Fetches the verified canonical amino acid sequence from UniProt (not AI-generated—this eliminates hallucination)
- Validates the sequence for correctness before proceeding
- Zero human intervention required
The Architect (Designer Engine):
- Enters an autonomous Actor-Critic design loop
- Generates protein mini-binder candidates (50–80 amino acids)
- Each candidate is critiqued using biophysics-informed rules:
- Hydrophobic core ratio (must be 20–60%)
- Charge balance (positive vs negative residues)
- Forbidden patterns (polyglycine, polyproline, charge clusters)
- Amino acid validity
- Invalid designs are rejected before structure prediction—saving compute time
- Continues iterating until a configurable number of candidates pass all gates.
The Physics Engine (Structure Prediction + Validation):
- Each accepted design is sent to Meta AI's ESMFold for 3D atomic structure prediction
- The predicted structure is validated using:
- pLDDT confidence score (extracted from the PDB B-factor column)—must exceed 70.0 to pass
- Radius of gyration—measures compactness
- Candidates with low-confidence structures are rejected automatically
- All passing structures are saved as PDB files
Molecular Docking (HDOCK):
- The top candidate structure is docked against the experimentally determined target protein structure (downloaded from RCSB PDB)
- HDOCK predicts how the designed binder physically binds to the target, producing:
- A docking score (binding strength estimate)
- A confidence score (reliability of prediction)
- Top 10 binding poses (different ways it could bind)
- Results are visualized as a docked complex
How I built it
Core Technologies
- Python—Pipeline orchestration, validation logic, agent coordination
- Google Gemini 2.0 Flash API—Fast iterative reasoning and design generation
- BioPython—FAST A parsing and sequence handling
- UniProt REST API—Ground-truth protein sequence retrieval (eliminates hallucination)
- Meta AI ESMFold—3D protein structure prediction
- HDOCK—Protein-protein docking and binding prediction
- PyMOL—3D structure visualization and validation
Agentic Design Logic Rather than asking an LLM for a single "best" answer, AEGIS enforces a self-critical reasoning loop:
- Generate a candidate sequence
- Validate it against physics-based rules (length, composition, forbidden patterns)
- Predict its 3D structure using ESMFold
- Check structure confidence (pLDDT)
- If it fails at any step, log the failure reason and iterate
- Only candidates passing ALL gates are accepted
This prevents hallucinated or meaningless outputs from entering the pipeline.
Challenges I ran into
The journey wasn't smooth. I faced three critical engineering hurdles:
- The Hallucination Problem:
Problem: Early versions would let the AI generate protein sequences directly. These sequences were often fabricated and scientifically meaningless. Solution: Sequences are now fetched exclusively from UniProt—a verified biological database. The AI is only used for identification (virus name, target protein), never for sequence generation. This is the single most important architectural decision in AEGIS.
- Windows Encoding Errors:
Problem: Writing output files on Windows failed because emoji characters and Unicode in reports were incompatible with the default cp1252 encoding. Solution: All file write operations explicitly use encoding='utf-8'. Emojis were removed from file outputs while keeping them in console output.
- JSON Serialization Failures:
Problem: NumPy float32 values from radius-of-gyration calculations caused json.dump() to crash, because JSON only supports native Python types. Solution: All numeric values are explicitly cast to native Python float() or int() before serialization. A dedicated type-conversion layer was added to the validator.
- Unreliable LLM JSON Output
Problem: Gemini sometimes returned conversational text mixed with JSON, breaking the parser. Solution: A custom extract_json() function uses regex to surgically extract valid JSON blocks from any mixed-text response.
Accomplishments that we're proud of
Test Target: SARS-CoV-2 Spike Glycoprotein
Genome: 29,903 base pairs (full SARS-CoV-2 genome) Target protein: Spike glycoprotein (1,273 amino acids) Source: UniProt ID A0A6B9XJC0 (verified, not AI-generated)
Design Results: The top-ranked model, Model_1, achieved the strongest binding with a docking score of –219.27 and the highest confidence score (0.7999), making it the most promising candidate despite a relatively high ligand RMSD of 348.71 Å. Model_2 and Model_3 followed with docking scores of –214.19 and –208.79, paired with solid confidence scores of 0.7831 and 0.7642. As the ranking progresses, docking scores become less negative (down to –187.36 for Model_10), and confidence scores gradually decrease from 0.7999 to 0.6786. Ligand RMSD values varied widely across models, ranging from ~311 Å to over 400 Å, reflecting differences in structural alignment. Overall, the analysis highlights Model_1 as the best candidate, with Models_2 and 3 also showing strong binding potential, while lower-ranked models exhibit weaker interactions.
AEGIS ran design iterations autonomously. Most candidates passed all validation gates:
One candidate (AEGIS_V001) was correctly rejected with pLDDT: 67.53 — visual inspection in PyMOL confirmed it was disordered and unstable. This validates that the pLDDT threshold is working correctly as a quality gate.
pLDDT interpretation:
- Above 90: Very high confidence
- 70–90: High confidence (all accepted candidates fall here)
- Below 70: Low confidence—rejected
3D Structure Validation (PyMOL) All accepted candidates were visually inspected in PyMOL. Every structure showed:
- Clean, well-formed alpha-helices
- Compact globular fold
- No disordered loops or structural breaks
The rejected candidate (V001) showed multiple disordered loops and an extended, unstable shape—confirming the automated rejection was correct.
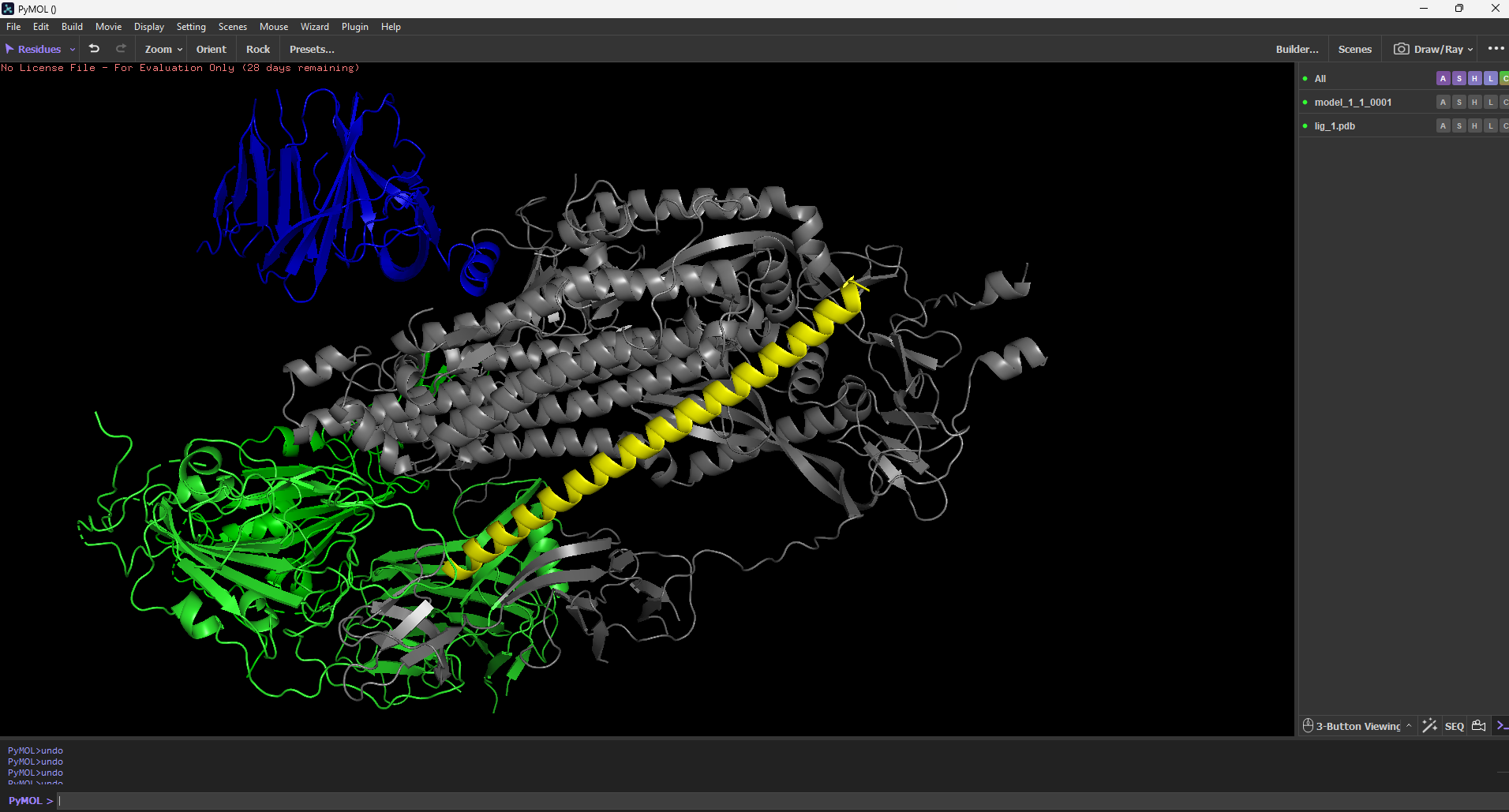
Molecular Docking Results (HDOCK) The top candidate (AEGIS_V006) was docked against the SARS-CoV-2 spike protein structure.
"AEGIS was run with expanded iterations (100 max, 10 candidates). The system correctly rejected 1 candidate with low pLDDT (67.53) and accepted 10 high-confidence structures. The top candidate, AEGIS_V007 (pLDDT 88.82), was docked to the SARS-CoV-2 spike protein (PDB: 6VXX) using HDOCK. Docking produced a score of -219.27 with 80% confidence, predicting binding at the trimer interface — a region targeted by some neutralizing antibodies. Visual inspection in PyMOL confirmed structural contact between V007 and the spike surface.
Safety & Physics Validation Layer
What AEGIS Checks Automatically
- Input validation — FASTA file is parsed and verified before any AI processing
- Sequence ground truth — Target sequences come from UniProt, not AI generation
- Design constraints — Candidates must pass hydrophobic ratio, charge balance, and pattern checks
- Structure confidence — pLDDT must exceed 70.0; low-confidence structures are rejected
- Compactness — Radius of gyration is calculated to verify compact folding
What AEGIS Does NOT Check (Honest Limitations):
- Actual binding affinity (docking scores are estimates, not measurements)
- Toxicity or immunogenicity (requires experimental assays)
- Whether the binding site is functionally relevant to blocking viral entry
- Manufacturability or stability in biological conditions
What I learned
AI as a reasoning engine, not just a text generator. By embedding Gemini inside a constrained pipeline with validation gates, AEGIS uses AI for what it does well (pattern recognition, sequence design) while preventing what it does poorly (hallucinating scientific data).
Fault tolerance is as important as capability. The most critical bugs were not about AI capability — they were about handling edge cases: encoding errors, type mismatches, malformed outputs. Production scientific software must be robust, not just smart.
Ground truth is non-negotiable. The single biggest improvement was replacing AI-generated sequences with UniProt-verified sequences. In scientific software, the data pipeline is everything.
Computational prediction is a hypothesis, not a conclusion. Docking scores and pLDDT are tools for prioritization, not proof. The value of AEGIS is that it generates testable hypotheses fast — not that it guarantees results.
What's next for AEGIS
- Integrate AutoDock Vina for local docking (gives kcal/mol, more standard)
- Benchmark AEGIS against known published binders for SARS-CoV-2
- Add comprehensive safety screening (BLAST against human proteome, immunogenicity prediction)
- Expand to other viral targets (Zika, Influenza, Ebola)
- Wet-lab validation: synthesize top candidates and measure binding experimentally
- Write methodology paper for bioRxiv
- Cloud deployment for researchers worldwide
- Expand to bacterial pathogens and oncology targets
- Integrate molecular dynamics simulation for stability prediction
Final Thought
" AEGIS is not a cure generator. It is a decision-compression engine. AEGIS does not replace scientists. It amplifies them. "
By shrinking the time between genome sequencing and scientifically credible, structurally validated hypotheses, AEGIS allows biology to begin sooner — when time matters most. The future of medicine is not AI replacing scientists. It is AI amplifying them.


Log in or sign up for Devpost to join the conversation.