-

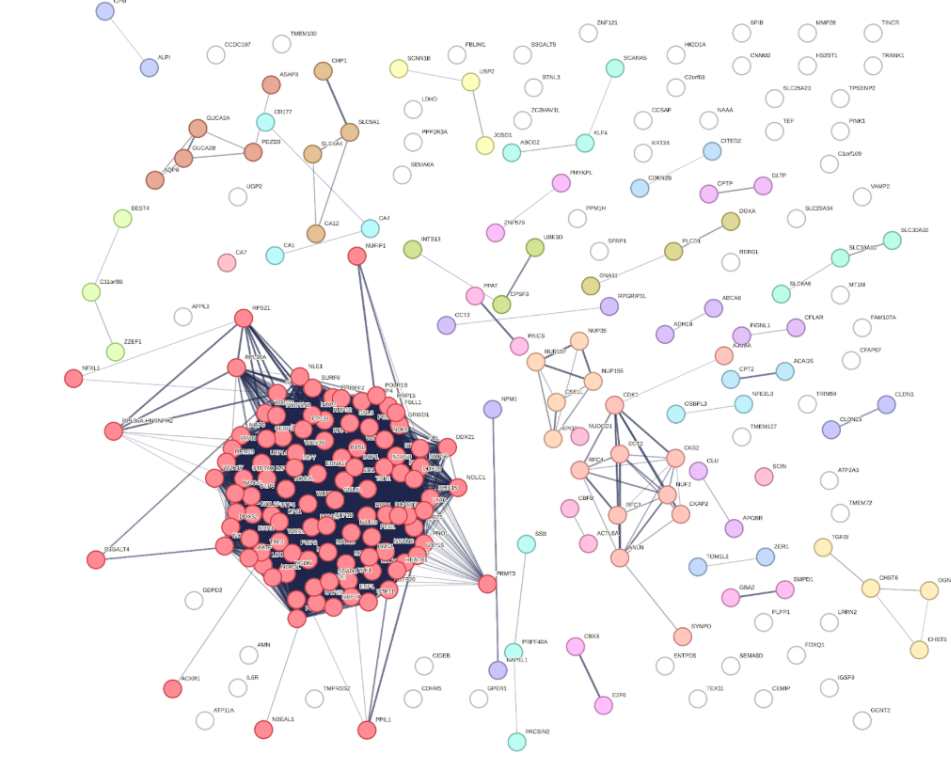
PPI network 1
-
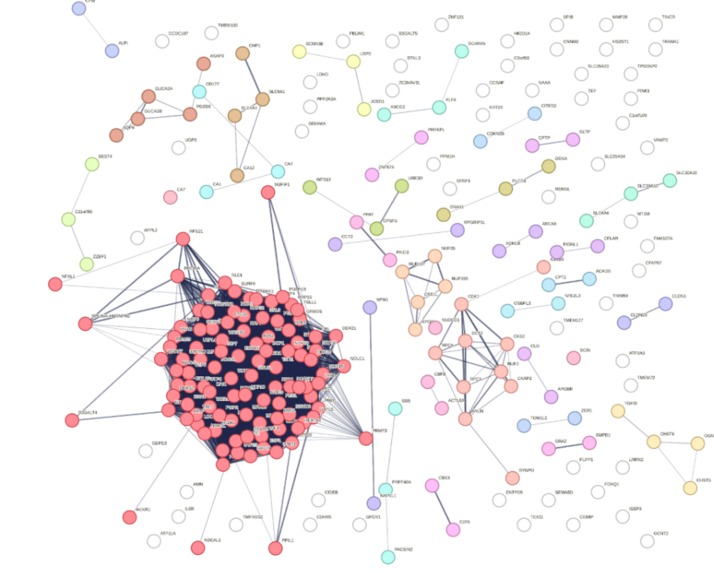
PPI network 2
-
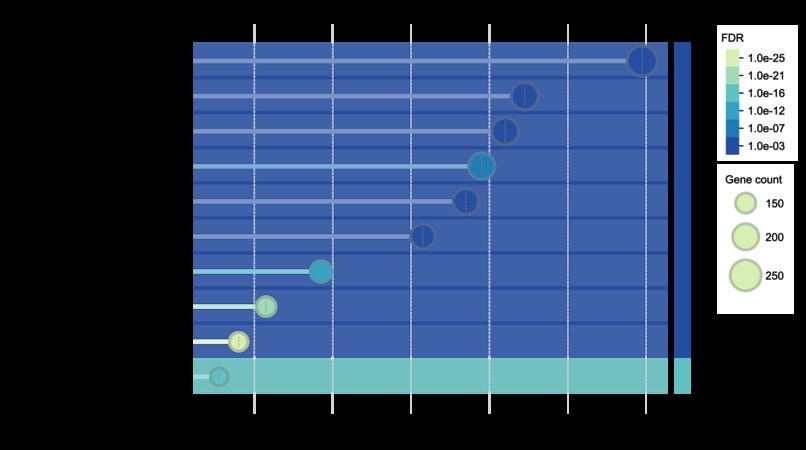
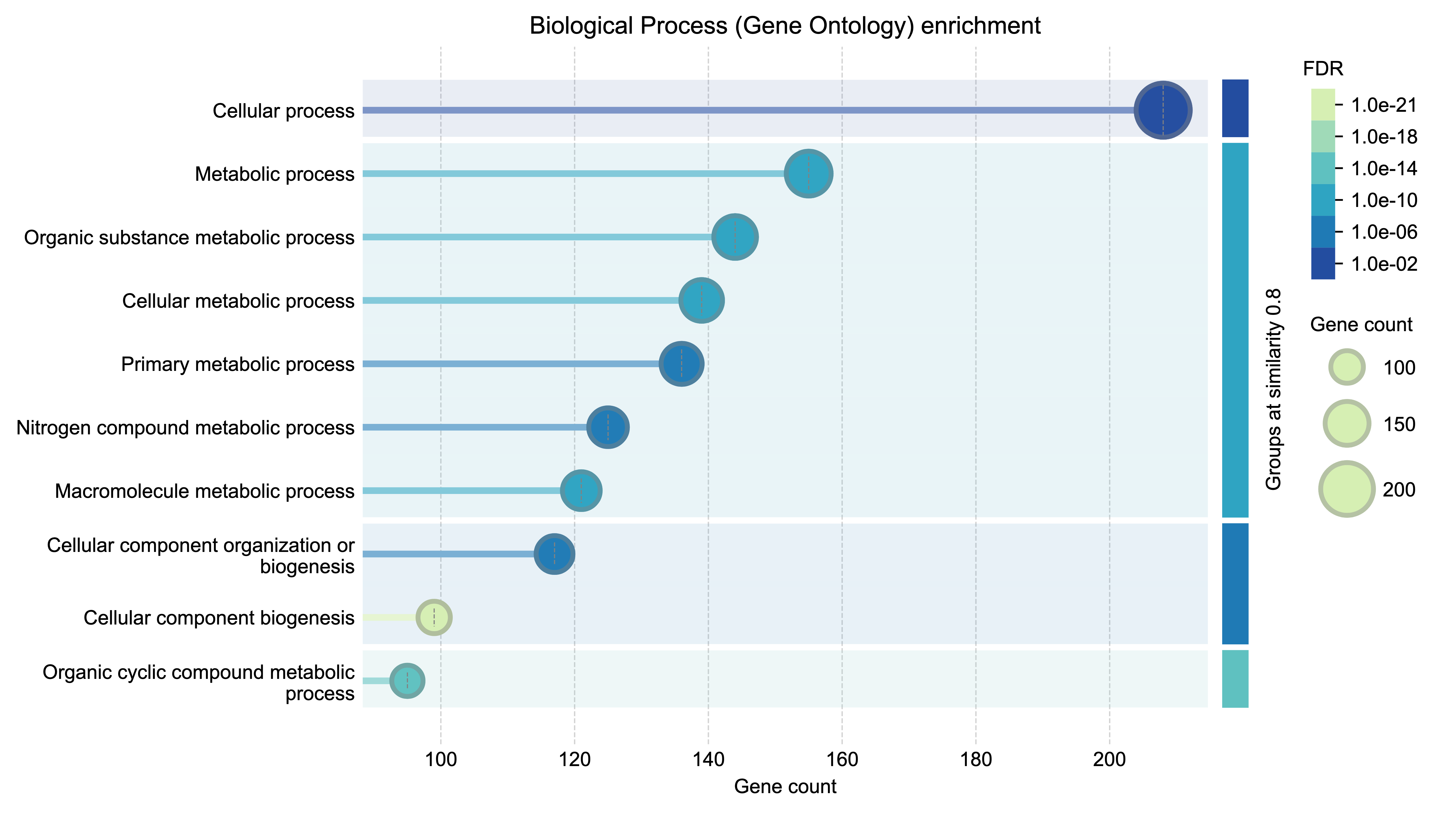
Functional enrichment analysis example 1
-
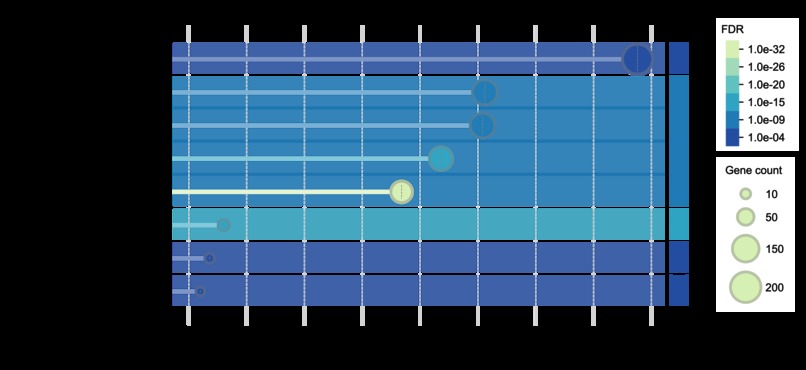
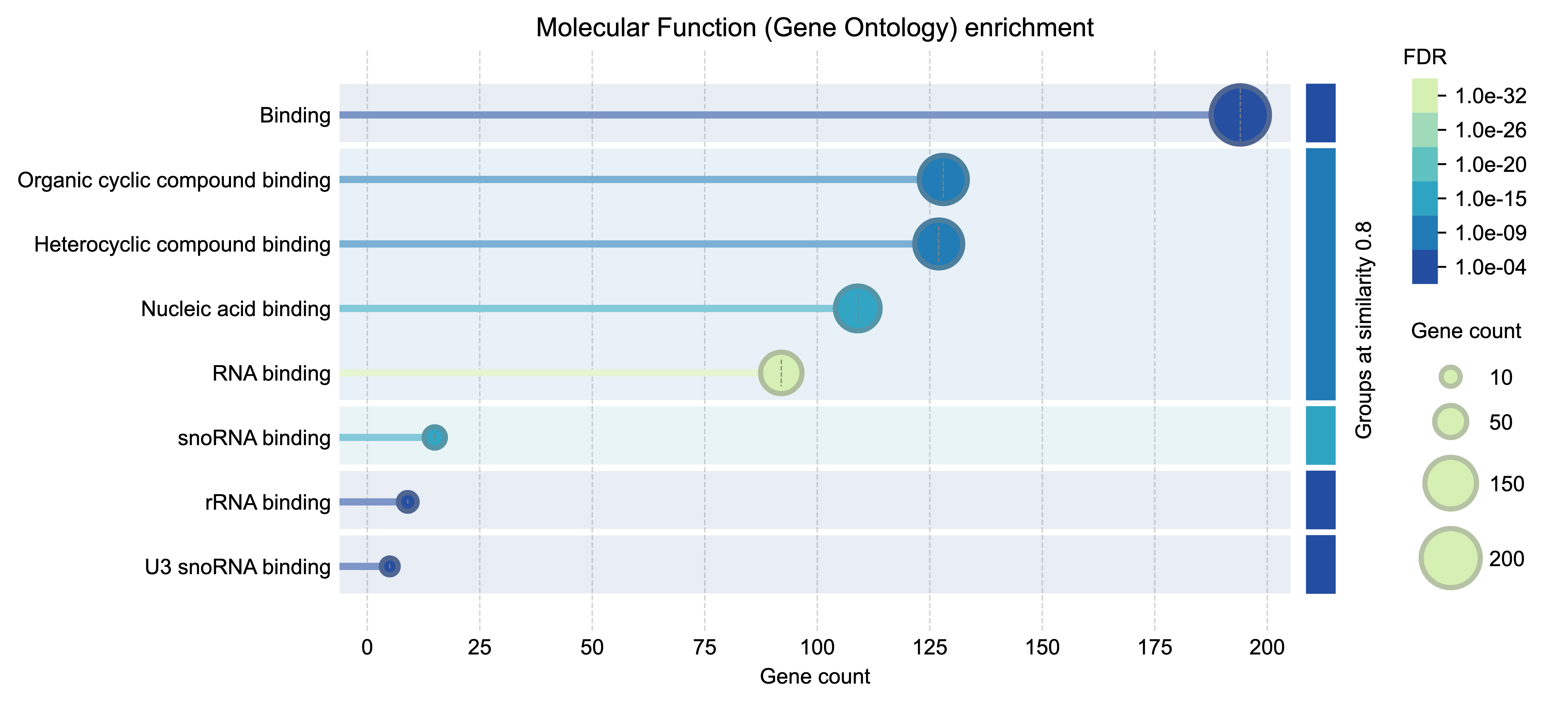
Functional enrichment analysis example 2
-
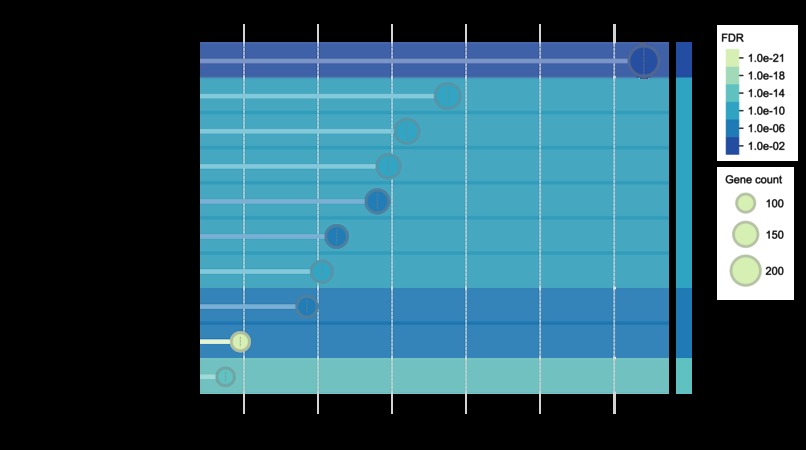
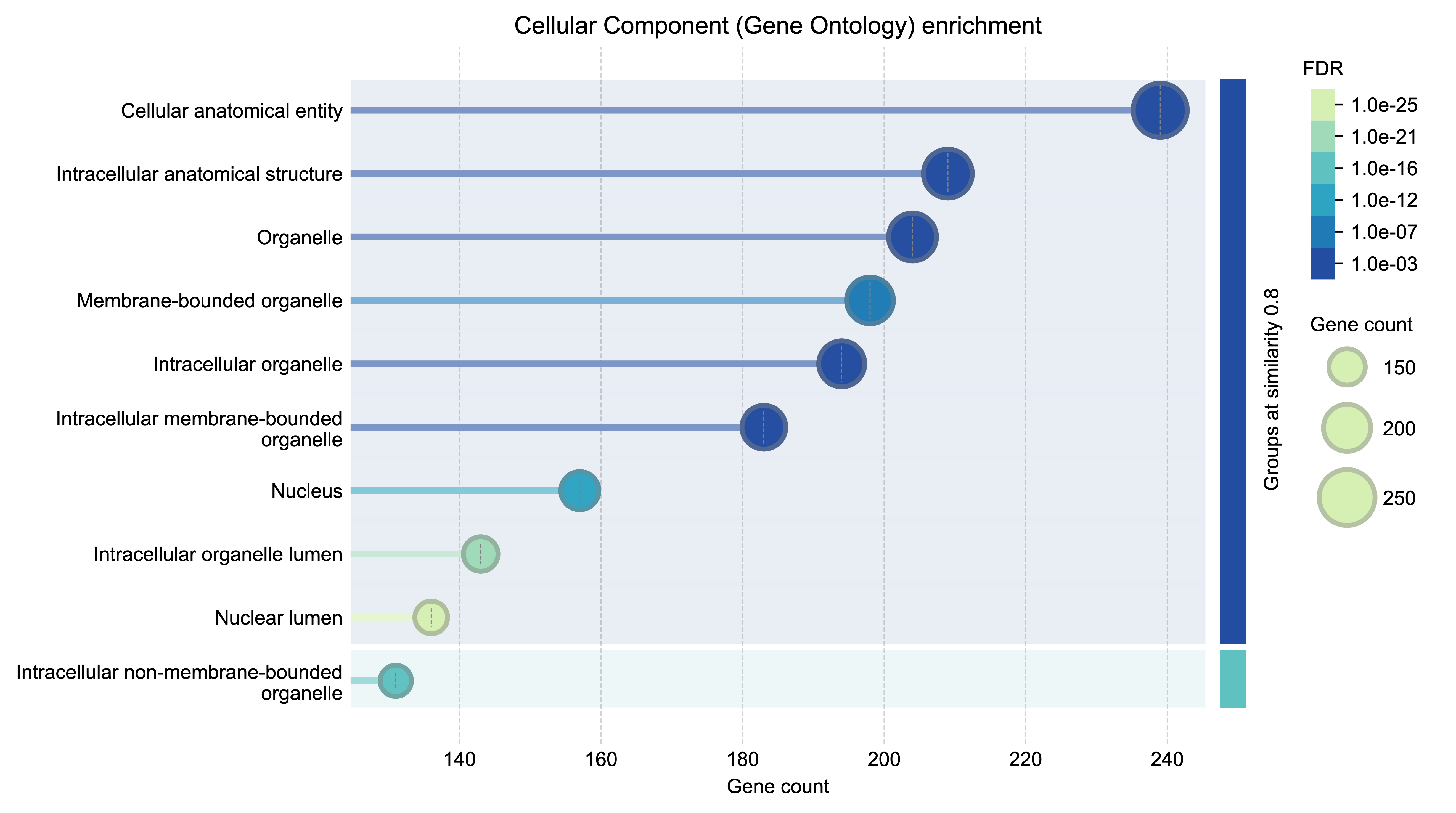
Functional enrichment analysis example 3
-
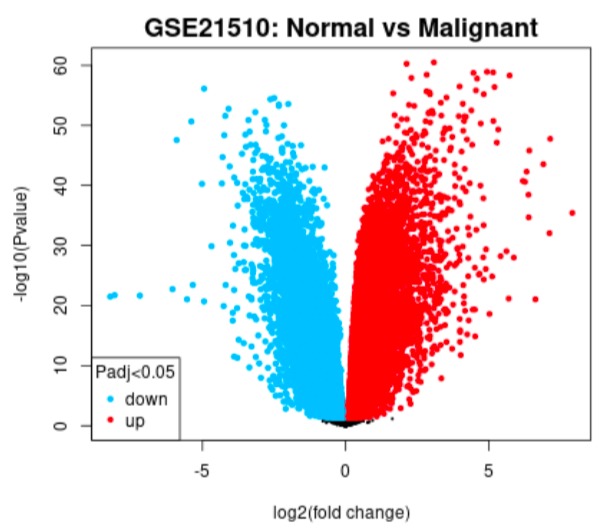
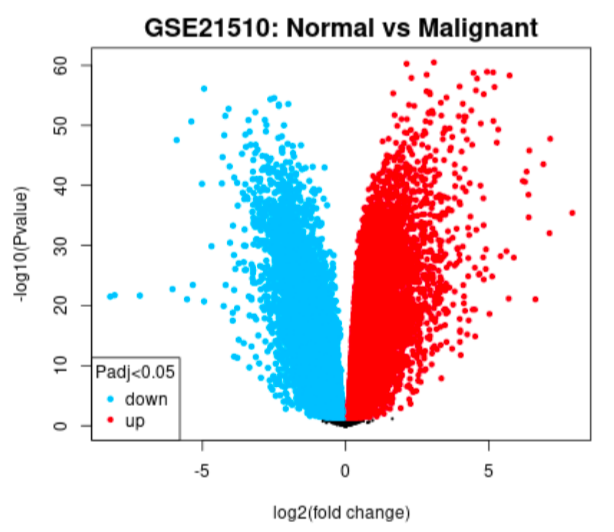
Volcano plot example
-
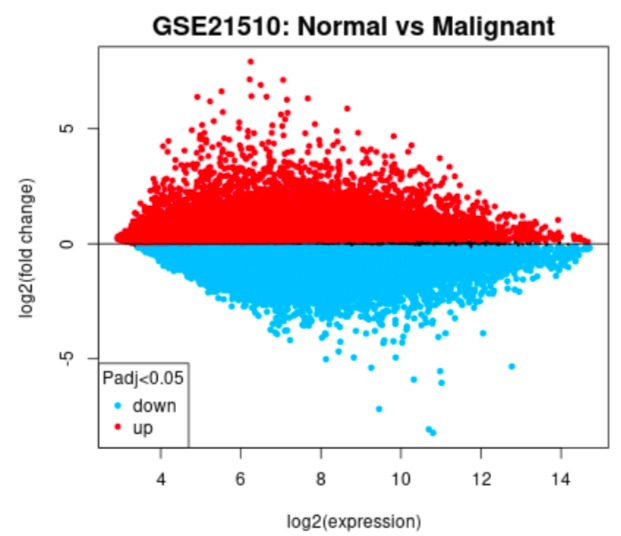
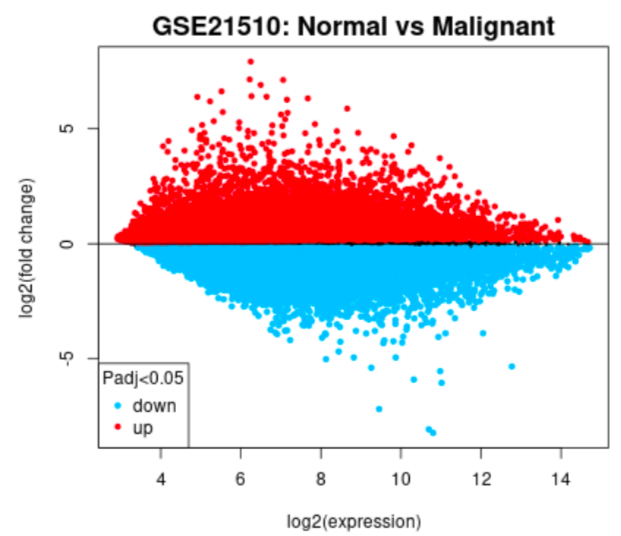
Main difference plot example
-
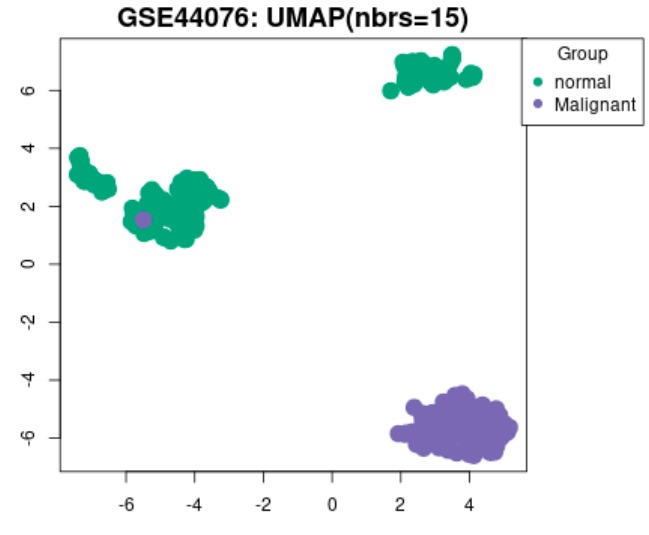
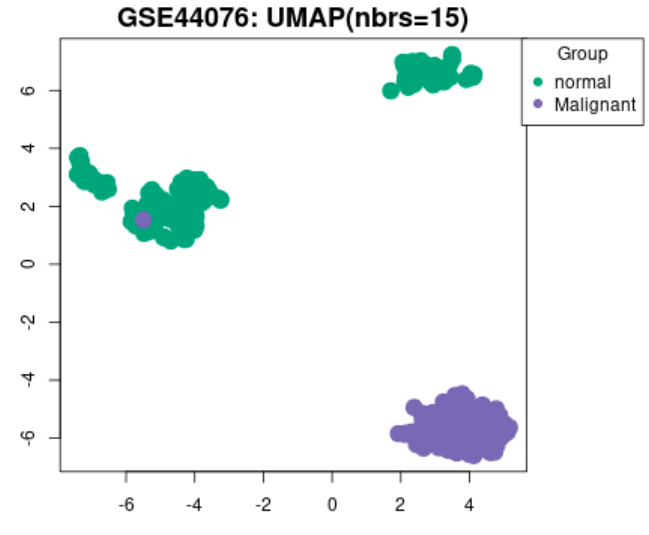
UMAP plot example

Inspiration
This project is inspired by recent work from a group of Korean scientists at the Korea Advanced Institute of Science & Technology (KAIST), who successfully reprogrammed malignant colon cells back to their normal state using digital twins of gene regulatory networks. This could significantly alter our approach to cancer therapies that do not use radiation, thereby reducing side effects. This sparked my interest in the fundamental principles of cell reprogramming and gene networks, as well as how they interact with each other.
What it does
This project provides a proof of concept of targeting genes for cell reprogramming using gene regulatory networks. I believe this prototype helps bring awareness to emerging cancer treatments and elevate students' engagement in biotechnology, as the method used in this prototype is accessible and is not based in a wet lab.
The gene regulatory networks we built allowed us to identify key hub genes (genes that have the most interactions, meaning they make a significant impact in the network) that can act as "switches" between malignancy and benignity. These were then highlighted in being potential targets for reprogramming.
How we built it
Publicly accessible tools are used: GEO(Gene Expression Omnibus is a database repository of high-throughput gene expression data and hybridization arrays, chips, microarrays), GEO2R ( a simple interface that allows users to perform sophisticated R-based analysis of GEO data to visualize gene mapping) and STRING (a biological database and web resource of known and predicted protein–protein interactions).
Our methods are inspired by a 2021 study on the progression of cervical cancer. The gene expression profile related to cancer progression was retrieved and downloaded from the Gene Expression Omnibus (GEO) database of the National Center for Biotechnology Information (NCBI). We have chosen GSE44076 and GSE21510 because they have been used to analyse hub genes by another group of scientists. We have also decided to standardize our selection by only using colon cells gene expression dataset, as inspired by KAIST’s work.
GEO2R tool was used to analyse the two datasets where we grouped the samples according to the information provided in the dataset (normal vs. cancer), and compare gene expressions to identify differentially expressed genes (DEGs).
The DEGs identified from GEO2R were entered into the STRING database to explore their potential protein-level interactions. We set the species to Homo sapiens and applied a medium confidence score cut-off of 0.4 to ensure reliable interactions while still capturing relevant connections. STRING generated a network of nodes (proteins) and edges (interactions), which was then exported as a table of interactions.
The dataset was then imported into Microsoft Excel, where the degree of connectivity for each protein was calculated using the COUNTIF formula by counting the number of interactions (edges) associated with each node. Proteins with the highest number of connections were considered hub genes as their high degree of interaction suggests an important regulatory role within the malignant gene network.
Challenges we ran into
One of the biggest obstacles was definitely navigating through GEO2R and STRING. Learning how to analyse the tables produced on GEO2R from the datasets such as volcano plots and UMAP plots was new and challenging. However, we managed to see the correlations between them and the data by researching relevant papers, which helped us integrate our results into our final write-up. Moreover, given both datasets were large, we had trouble including all of the data in the STRING network. Luckily, we decided to reduce the number of genes in each network by only including the top 250 DEGs. This way, our methods were standardized.
Accomplishments that we're proud of
We managed to complete our project and identify our hub genes for reprogramming, which was our final goal. Moreover, being able to reflect on it by addressing the limitations of our project was also a great achievement for us, as we know how our in-silico method is restricted compared to a wet-lab one, providing future directions for our project.
What we learned
Overall, we learnt about the fundamental principles of cell reprogramming and how it is a promising approach to cancer therapies by reducing side effects such as radiation. Most importantly, we gained knowledge in bioinformatics by navigating through gene analysis and mapping, which is a valuable skill to have in the biotechnology industry.
What's next for "Addressing cancer therapy: a protoype of gene analysis"
Through research, we gathered potential future wet-lab approaches to our project such as CRISPR, AI models and a personalised medicine approach. By addressing these next steps, the promising predictions of this prototype can be rigorously tested, refined, and translated into a tangible strategy for overcoming cancer through network reprogramming.


Log in or sign up for Devpost to join the conversation.