-
-
Breite der Pfeile und Boxen sind proportional zur relativen Häufigkeit
-
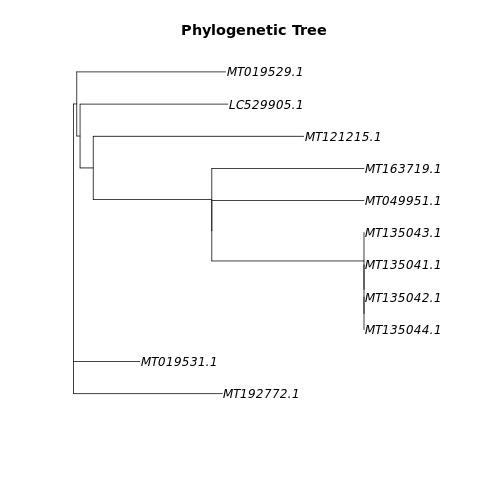
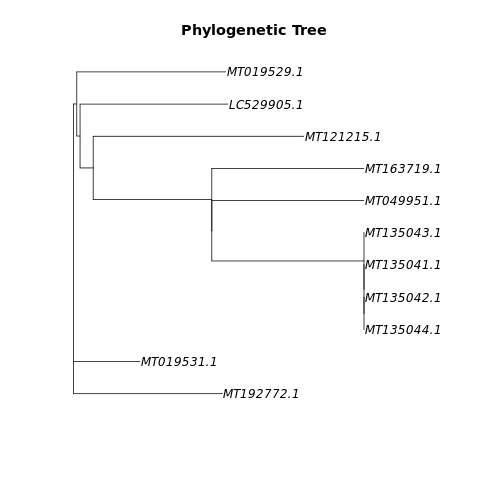
Phylogenetischer Baum erzeugt aus unserer Beispielpipeline
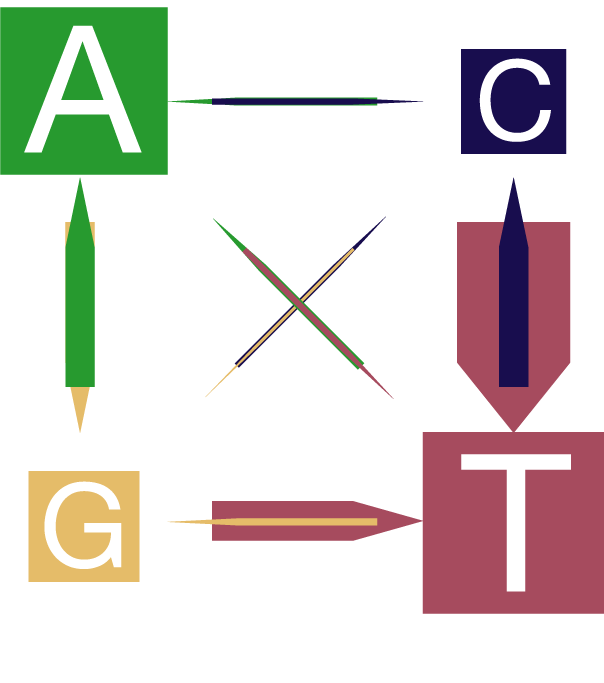
Inspiration
What it does
The available phylogenetic data by nextrain.org, which are based on GISAID (www.gisaid.org) suggests that single nucleotide polymorphisms occur anywhere on the RNA. In view of the importance of new virus strains, the aim of the project is to use data science on genome and mutation data to find patterns in mutations and enable predictions therefrom. Specifically, build a workflow and tools to find patterns in the nextstrain data set.
How we built it
We downloaded the SARS-CoV2 population data from Nextrain in JSON format and turned it into a dataframe for analysis. We used Python and a Jupyter notebook for statistical analysis. We included genome data from NCBI and in our calculation of variant positions. We recognize that we tapped only a small fraction of all available genome data. For this task different toolsets and languages were used, dockerfiles installing all dependencies are available.
Challenges we ran into
The mutation data, wonderfully visualized as it is by Nextrain, falls short in several aspects as primary data for big data research.
1) It is a summary of sequencing data after alignment for the purpose of virus strands and the epidemic dynamics. It is not a full set of primary data. For example, the neighbouring nucleotides need to be reconstructed. Not difficult conceptually but time-consuming programming.
2) The set of mutations is rather small for statistical inference. The virus comes with roughly 30 000 nucleotides whereas we have roughly only 1000 independent mutations throughout the whole population.
3) Retrieving data from public databases, constructing mutation positions and phylogenetic dependencies is not a task that is done fast. Most of the datasets and calculations have different output formats. To bring them into a merged structure for a data processing workflow needs not only programming efforts but also knowledge of evolutionary mechanisms.
Virus sequencing and the understanding of mutations both in terms of understanding the epidemic, the development of vaccines, and its microbiology is obviously a challenging subject. The team tried its best to find a quick path into the subject but we have to concede that this project did not end in the sense defined at the outset.
The team split into different groups. We might have benefited from more microbiological input and direction as well as concrete tasks.
Accomplishments that we are proud of
Within a weekends time do meaningful statistics on viral genome data without any prior exposition. Get into this subject of bioinformatics, read various publications and scholarly research. In a very limited time, find a way into this highly relevant subject of the corona virus’ genome and mutations. In addition, we can provide workflows based on open-source software to gather sequences and information from several sources, process alignments and extract variants and finally construct a phylogenetic tree. This might be useful to compare COVID19 to other organisms and to gain phylogenetic information.
What we learned
Single nucleotide mutations are characterized by a position on the RNA strand and nucleotide context. Viewing the sequencing data as a sample of independent observations, we observe the following statistics:
1) Mutations are not uniformly distributed by position. They are not very clustered in the sense that they are concentrated on small regions.
2) Also in the original genome, nucleotides themselves are not equally distributed, which affects the mutation types that may happen there
3) There are significant differences in the frequencies of single nucleotide conversion pairs. The genome is already rich in Ts and through the mutations, the occurence of this nucleotide is further increased. (The width of the arrows and boxes is proportional to the frequencies of the conversion and nucleotides, respectively). Figure above or under this link:
4) An attempt to explain these differences by sequence patterns in the neighbourhood of a mutation remained incomplete.
What's next for 1_041_a_forschung Mutationsvorhersage
Eine Fortsetzung dieser Arbeiten ist denkbar. Dazu bedürfte es allerdings einer gewissen Koordination und der Mitarbeit einer wissenschaftlichen Institution in diesem Gebiet. Die entwickelten Verarbeitungsplattformen können dabei als erste Grundlage dienen und eine technische Basis bereitstellen. Weiterhin wird ein bestehendes Forschungsprojekt an Mutagenesemechanismen von Covid-19 durch die Entwicklungen unterstützt.
Discord
https://discordapp.com/channels/690647716274896978/690689583272296539
Built With
- docker
- java
- javascript
- jupyter
- python
- r











{kind=link}
Log in or sign up for Devpost to join the conversation.